
SiGN-BN
1. プログラム名
SiGN-BN
2. 開発責任者
宮野 悟(東京大学 医科学研究所)
3. 主な開発者
井元 清哉(東京大学 医科学研究所)
玉田 嘉紀(東京大学大学院情報理工学系研究科)
4. 内容
概要
ベイジアンネットワーク・モデルを用いて細胞内分子の発現制御システム(遺伝子ネットワーク)のモデル化・予測を行う超並列計算機用ソフトウェア群。
詳細
次世代シークエンサーやDNAチップなどの計測技術を用いて得られた細胞内遺伝子発現量の多種類のデータ(遺伝子発現データ)から遺伝子間の統計的因果関係を推定するプログラムである。
相関関係を統計的因果関係の代替変数にみなすことが安易になされることもあるが、統計学的には「相関関係がある」ことは「因果関係がある」ことを意味しない。ベイジアンネットワーク・モデルは最も厳密に統計的因果関係を推計する方法である。計算結果は遺伝子を節点(ノード)、遺伝子間の制御関係を節点間の有向枝(エッジ)とする有向グラフとして表示される。例えば「遺伝子A→遺伝子B」と表示されると、遺伝子Bは遺伝子Aによる制御を受けていると推測される。実験計画の段階からあらかじめ注目する遺伝子が存在する場合、その遺伝子と有向枝で結ばれる遺伝子がベイジアンネットワークによって統計的因果関係のある遺伝子として浮かび上がってくる。また、多くの遺伝子と統計的因果関係を持つ遺伝子はハブ遺伝子と言われ、研究対象である生命現象の鍵遺伝子であると推測されている。
遺伝子発現データは個人の細胞サンプルや遺伝子ノックダウン実験,薬剤投与による時系列データなどを用いることが可能である。時系列データに対しては動的(ダイナミック)ベイジアンネットワークを、ノックダウン実験などの静的なデータに対しては通常のベイジアンネットワークを用いて遺伝子ネットワークを推定する。
(参照:http://sign.hgc.jp/signbn/)
5. どんなことができるか
- ヒト全遺伝子産物を対象にした遺伝子ネットワーク推定による創薬ターゲット遺伝子のイン・シリコ探索が大規模にできる。
- ネットワークを用いて、被影響遺伝子の同定、作用点の推定、副作用の予測・回避、創薬ターゲット・毒性関与パスウェイ探索などが大規模に行える。
- 様々なデータから得られる様々な遺伝子ネットワークを超短時間で計算できるようになる。
6. 関係論文
[1] Imoto, S., Goto, T., and Miyano, S. (2002). Estimation of genetic networks and functional structures between genes by using Bayesian network and nonparametric regression. Pacific Symposium on Biocomputing, 7, 175-186.
[2] Tamada, Y., Shimamura, T., Yamaguchi, R., Imoto, S., Nagasaki, M., and Miyano, S. (2011). SiGN: Large-scale gene network estimation environment for high performance computing, Genome Informatics, 25 (1), 40-52. [Pubmed]
[3] Tamada, Y., Imoto, S., Araki, H., Nagasaki, M., Print, C., Charnock-Jones, D.S., and Miyano, S. (2011). Estimating genome-wide gene networks using nonparametric Bayesian network models on massively parallel computers, IEEE/ACM Transactions on Computational Biology and Bioinformatics, 8 (3), 683-697. [Pubmed]
[4] Tamada, Y., Imoto, S., and Miyano, S. (2011). Parallel algorithm for learning optimal Bayesian network structure, Journal of Machine Learning Research, 12, 2437-2459. [Publisher]
7. チュートリアル資料
- 玉田 嘉紀 (2011)「大規模遺伝子ネットワーク推定ソフトウェアSiGN」
- 戦略分野1 (2013)「SiGN講習会資料:ベイジアンネットワークを用いた遺伝子ネットワークの推定と解析」
- 戦略分野1 (2013)「SiGN-BN 実習テキスト」
- その他利用者向けチュートリアル「利用者向けチュートリアル」を参照ください。
8. 関連する教科書等
井元 清哉 (2008) 生命システムネットワークを明らかにするための統計的モデリング,(統計学会75周年記念出版,「21世紀の統計科学II 自然・生物・健康の統計科学」小西 貞則,国友 直人 (編)),東京大学出版会,153-180.
9. マニュアル
利用者マニュアルを参照ください。
10. 処理の手順
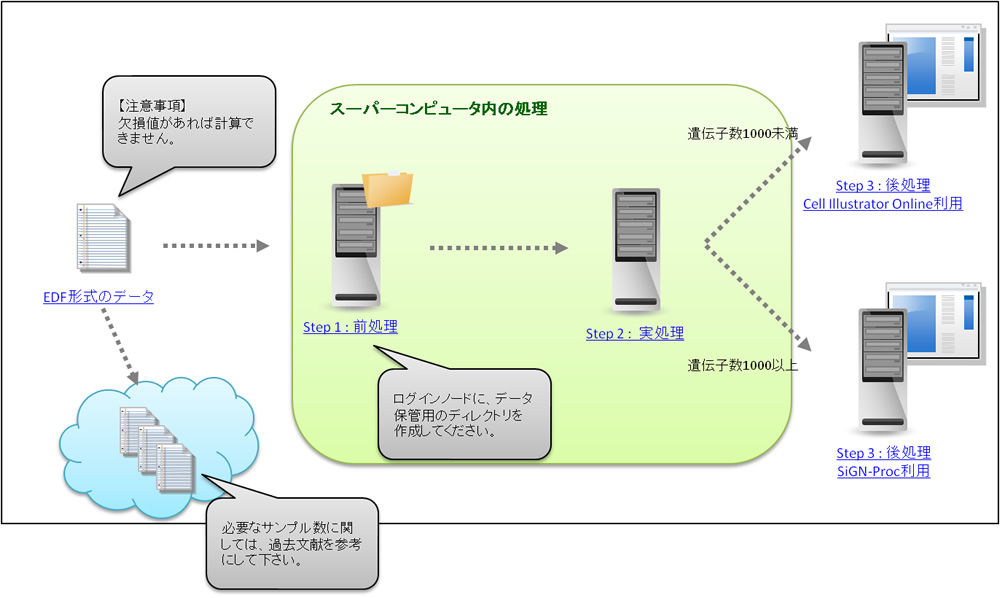
処理方法の詳細は(http://sign.hgc.jp/signbn/howto.html)を参照ください。
入力データ
SiGN-BNによって遺伝子ネットワークを推定するためには、まずEDF形式(Expression Data Format)の遺伝子発現データが用意されなければならない。EDF形式の詳細はマニュアルを参照のこと。また、遺伝子発現データには欠失値が含まれていてはならない。
どれほどの実験回数の遺伝子発現データセットが必要かを示すことは困難である。関連論文を参照されたい。
処理
SiGN-BNでは遺伝子ネットワーク推定のために、HC+Bootstrapアルゴリズムと、NNSRアルゴリズム、Para-OSアルゴリズムが用意されている。(詳細はマニュアルを参照されたい。)われわれは多くの研究でHC+Bootstrapアルゴリズムを用いているが、この方法では1,000程度の遺伝子しか扱えない。NNSRアルゴリズムでは20,000以上の遺伝子を処理できる。Para-OSアルゴリズムはベイジアンネットワークでいう最適なスコアをもつベイジアンネットワークを推定しうるが、取り扱える遺伝子数は32以下である。
後処理
処理が終了するとCSML形式やテキスト形式の遺伝子ネットワークが得られる。CSML形式のデータはCIO(Cell Illustrator Online)を用いてネットワーク表示される。しかし、CIOでは1,000以上の遺伝子からなるネットワークを取り扱うのは困難である。1,000以上の遺伝子を取り扱う場合には、興味ある遺伝子の周囲の部分ネットワークを取り出したり加工・表示できるプログラムSiGN-Procを用いるのがいい。
11. ソフトウェアのダウンロード
ソフトウェアはこちらからダウンロードできます。
(1) スーパーコンピュータ「京」での実行
スーパーコンピュータ「京」での実行環境も整えています。利用ご希望の方は、主な開発者である玉田 嘉紀(東京大学大学院情報理工学系研究科)までご連絡ください。
(2) SCLS計算機システムで実行するソフトウェア
SCLS計算機システムでの実行環境も整えています。利用ご希望の方は、SCLS計算機システム![]() までご連絡ください。
までご連絡ください。
12. 利用許諾権(ライセンス)について
SiGN-BNはオープンソース・ソフトウェアではありません。ダウンロードしたバイナリーファイルの再配布は禁止です。
SiGN-BN is not the open source software. DO NOT re-distribute the binary file.
上記情報は2013年7月5日のものです。最新の情報はSiGN-BNのホームページでご確認ください。
13. ソフトウェアの詳細
(1) 計算モデル化の方法
ノンパラメトリック回帰ベイジアンネットワーク
(2) 計算方法
発見的構造推定アルゴリズム+ブートストラップ法、Neighbor Node Sampling & Repeatアルゴリズム、並列最適構造推定アルゴリズム
(3) 並列化の方法
MPI,OpenMP
(4) 開発言語等
C
(5) スーパーコンピュータ「京」での最大計算規模
全転写産物(10万以上)の遺伝子ネットワーク推定
- 64万コアを使用
- メモリ容量 1PB、ディスク容量 10GB/network







