
MP-CAFEE
1. プログラム名
MP-CAFEE
2. 開発責任者
藤谷 秀章(東京大学 先端科学技術研究センター 特任教授)
3. 主な開発者
藤谷 秀章(東京大学 先端科学技術研究センター 特任教授)
山下 雄史(東京大学 先端科学技術研究センター 特任准教授)
4. 内容
概要
超並列アルゴリズムを用いた分子動力学シミュレーションで、タンパク質とリガンドの結合自由エネルギーを高精度に計算します。
詳細
タンパク質PとリガンドLの結合自由エネルギー⊿Gbindは次に示す3つの反応過程
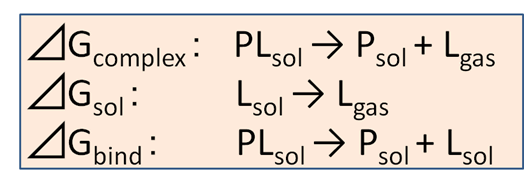
図1 ⊿Gbindを求めるための3つの反応過程
から次式
⊿Gbind=⊿Gcomplex-⊿Gsol (1)
で求めます。すなわち、⊿Gbindを求めることは、図1の最初の2つの反応過程を分子動力学計算でシミュレーションし、⊿Gcomplexと⊿Gsolを求めることに帰着されます。その反応過程をシミュレーションする分子動力学計算では、リガンドL内部の相互作用は変化させないで、リガンドと標的タンパク質Pおよび水など他の分子との相互作用を、等温等圧下で消滅させていきます。
⊿Gcomplexと⊿Gsolなど⊿Gの計算にはJarzynskiの関係式
![]() (2)
(2)
を用います。ここで系Aと系Bは図1の反応過程の左辺と右辺に対応します。Jarzynskiの関係式は、ポテンシャル関数UA(x)なる系AからUB(x)なる系Bに変化させ、その間になされた仕事量をWA→Bとすると、式(2)の右辺から自由エネルギー変化⊿Gが求まることを示しています。ここで<>A→Bは、A→Bへの変化を多数回試み、そのアンサンブル平均をとることを示しています。
図1の右辺の構造は比較的容易に設定することができますが、複合体PLsolの構造を設定することは容易なことではありません。このことを踏まえると、Jarzynskiの関係式で重要なことは、
・初期の系であるAは平衡状態になければならない。
・A→Bへの切り替えの経路や時間は問わない。
の2点です。一点目は共結晶構造やドッキングシミュレーションで得た構造を初期構造として平衡化のための分子動力学計算を行うことによって対応されます。二点目から系Aから系Bへの変化を瞬時に行うとするとJarzynskiの関係式(1)は
![]() (3)
(3)
となります。この場合、A→Bの間に中間状態はなく、UB(x)はA→Bへの変化時の系Aの状態xを用いて計算されます。実際には、系Aの分子動力学計算を行いつつ、ある時刻の状態xtを用いて、UA(xt)とUB(xt)の値を計算します。
MP-CAFEEでは系AからBへの変化を段階的に行わせることにより、結合自由エネルギーを高精度に求めています。図1の最初の反応過程に則して考えると、図2がその概念図です。
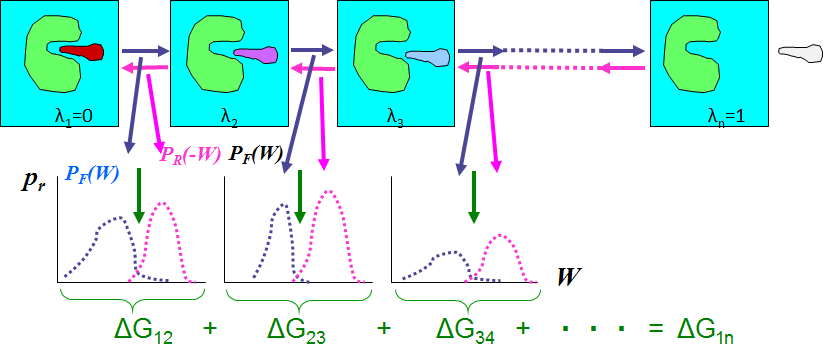
図2 MP-CAFEEの概念図
MP-CAFEEではλnのnを32としています。そして系λiのポテンシャル関数のクーロン力とファンデアワールス力の部分(ソフトコアポテンシャル)を次式(4)で定めています。
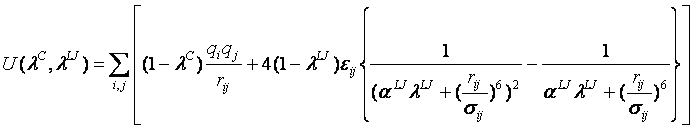 (4)
(4)
そして、MP-CAFEEでは、まず、クーロン相互作用を(λCを0→1とすることにより)断ち切り、次いでファン・デル・ワールス相互作用を(λLJを0→1とすることにより)断ち切ります。この順序を逆にすると、正と負の荷電粒子が急接近し、極めて不安定な状態が生じるため、順序は重要です。また、λCとλLJはそれぞれ、11個のλC(0.0, 0.1, 0.25, 0.4, 0.55, 0.65, 0.725, 0.8, 0.875, 0.95, 1.0)と21個のλLJ(0.0, 0.1, 0.2, 0.3, 0.4, 0.475, 0.55, 0.6, 0.65, 0.675, 0.7, 0.725, 0.75, 0.775, 0.8, 0.825, 0.85, 0.875, 0.9, 0.95, 1.0)、合計で32点からなっています。これが先ほど述べたn=32に対応します。その変化を図3に示します。
a)λCの変化
![]()
b)λLJの変化
![]()
図3 ソフトポテンシャルを決めるλ
MP-CAFEEで結合自由エネルギーを求める実行手順を以下に示します。
1)λ1の状態(標的タンパク質とリガンドが複合体を形成している状態)の平衡状態化を行う。
2)その平衡状態x0をすべてのλi(32点)での初期状態とする。
3)すべてのλiで12種類の初期運動量(T, P一定というアンサンブルに従った運動量分布)を生成し、分子動力学計算を行う。合計32×12の分子動力学計算を行うことになる。
4)あるλiでの分子動力学計算を行い、時刻tjでの位置座標を,xtjを用い、U(λi-1,xtj)、U(λi,xtj)、U(λi+1,xtj)を計算する。
5)このU(λi,xtj)、U(λi+1,xtj)を用いて、Jarzynskiの関係式から結合自由エネルギーを計算する。
6)MP-CAFEEではU(λi-1,xtj)、U(λi,xtj)、U(λi+1,xtj)を用いて、Acceptance Ratio法で結合自由エネルギーを求めている。なお、Acceptance Ratio法はGROMACSに組み込まれている。
5. どんなことができるか
- ある標的タンパク質を対象に設計された多くのリガンド(候補薬物)が標的タンパク質と結合する過程での自由エネルギー変化を求め、結合の強度を定量的に推計する。
- FKBPとFK506などのリガンドを例とした計算でMP-CAFEEが精度よく結合自由エネルギーを計算しうることを実証した。
6. 関係論文
[1] H. Fujitani, Y. Tanida, M. Ito, G. Jayachandran, C. D. Snow, M. R. Shirts, E. J. Sorin and V. S. Pande: “Direct calculation of the binding free energies of FKBP ligands” J. Chem. Phys. 123, 084108-5 (2005)
[2] H. Fujitani, Y. Tanida and A. Matsuura:“Massively parallel computation of absolute binding free energy with well-equilibrated states”, Phys. Rev. E, 79, 021914 (2009).
なお,田辺三菱製薬㈱の研究者によって下記論文が発表されています。
[3] Okada O, Yamashita H, Takedomi K, Ono S, Sunada S, Kubodera H. (2013) Prediction of the binding affinity of compounds with diverse scaffolds by MP-CAFEE, Biophys Chem. 180-181:119-26
[4]Hideaki Fujitani, Keiko Shinoda, Takefumi Yamashita, and Tatsuhiko Kodama(2013) High performance computing for drug development on K computer, J. Phys.: Conf. Ser. 454 012018
7. チュートリアル資料など
[1]藤谷秀章“分子動力学による結合自由エネルギー計算”, 日本薬学会 SAR News No.17, 2-8 (2009)
8. 関連する教科書
[1] M.P. Allen and D.J. Tildesley, Computer Simulation of Liquids (Oxford)
[2] D. Frenkel and B. Smit Understanding Molecular Simulation: From Algorithm to Applications (2nd Ed) (Academic Press)
9. 問い合わせ先
MP-CAFEEに関するお問い合せは、SCLS計算機システム![]() までご連絡ください。
までご連絡ください。







