
要旨
- 分子シミュレーション専用計算機の開発:過去と未来
- タンパク質-化合物複合体の量子化学計算-酵素反応の解明と創薬への応用に向かって-
- 医薬品設計のための大規模分子シミュレーション
- 系統樹解析と分子動力学計算に基づく[NiFeSe]ヒドロゲナーゼの分子進化の3Dシミュレーション
- 数学と大規模シミュレーションから読み解くがんの自然史
- 国際連携による癌の全ゲノムシークエンスとスーパーコンピュータ
分子シミュレーション専用計算機の開発:過去と未来
泰地 真弘人
理化学研究所 生命システム研究センター
我々はこれまで、分子動力学計算のための専用計算機の開発を継続的に行ってきており、現在その第4世代機「MDGRAPE-4」の開発を進めている。フォールディング等に代表されるタンパク質の大きな構造変化はしばしばミリ秒以上のゆっくりした時間スケールでおこる。一方、原子レベルでの運動の基本的な時間スケールはフェムト秒単位であり、ミリ秒でのイベントを観測するにはその間に横たわる1,000 兆にわたる時間スケール差を埋める必要がある。状態遷移モデルに基づく方法など、この差を埋めるために色々な手法が考案されてはいるが、単純にシミュレーションするとして1,000兆ステップを数ヶ月で実行するためには、1ステップを実時間数10マイクロ秒で実行できればよい。京コンピュータなどの汎用並列計算機は、演算能力としては十分にこれを満たす能力があるのだが、実際には並列化の限界から1ステップに数ミリ秒かかり、要求性能に比べて100倍遅い。実は前世代の専用計算機MDGRAPE-3(2006年完成)においても同じ問題があり、系を大きくした場合の性能向上「弱スケーリング」には問題なかったが、系の大きさを一定に保ったまま時間スケールを延ばす「強スケーリング」は実現できていなかった。通信等について専用計算機を接続するホスト計算機に依存しており、結局そこでの遅延がボトルネックになるためである。
その後米国D. E. Shaw Researchで2008年に開発された専用計算機Anton[1]では、汎用計算コア・専用計算機・ネットワークを統合した専用のシステムオンチップ(SoC)を採用することでこの問題を解決し、1ステップ約20マイクロ秒の非常に高い性能を得ることに成功した。彼らの専用部分はMDGRAPEと類似しているが、他部分との統合が大きく性能に影響している。この間にSoC開発のための設計資源が整ってきたことも大きい。
そこで我々も彼らに追いつくべく、SoCベースの専用計算機MDGRAPE-4の開発を2010年から開始した[2]。基本的にはこれまでのMDGRAPEでのホスト計算機まで含めた部分を1つのLSIに納め、これを並列化することによって高性能を目指している。また高速な同期のためにメッセージ送受信機能やメモリでの累積演算機能を備えるなど、分子動力学計算の高速化に向けた機能を有する。現在ハードウェアを完成させ、ソフトウェア(GROMACS)の移植を進めているところである。
このような専用計算機の方法は、今後も更に有効であると考えられる。現在、CPUの動作周波数の向上は非常にゆっくりであり、計算機の性能向上は殆ど全て並列度の向上に依存している。エクサスケールでは、実に億オーダーの演算器の数を持つことになる。しかし、この並列度の上昇を実効性能の向上に結びつける手段はそう多くなく、結局は通信やメモリアクセス等計算機の「足腰」の部分を強くする必要がある。専用化は低コストで足腰の強化を実現できる一つの方向性であり、ムーアの法則の終焉を見据えて今後より取り組みを進める必要があると考えられる。

図(a) MDGRAPE-4システムの全体写真。全部で512個のLSIを64個の筐体に搭載しており、全体で4ラックとコンパクトである。消費電力は約70kWである。オレンジ色のケーブルは筐体間の接続に用いている48芯の光ファイバケーブルで、全部で18,432芯のファイバを用いている。(b) MDGRAPE-4のシステムボードの写真。8個のLSIと48個の光送受信モジュールを搭載している。(c) MDGRAPE-4 LSI (SoC)のイメージ図。上下のPPが専用計算部分、左右のGPが汎用計算部分、中央部のGMがメモリ、中心のCGPが制御プロセッサ、周囲のNIFがネットワーク。その他命令メモリ(IMEM)、ホストインターフェイス(FPGA IF)がある。
講演では、これまでの専用計算機開発の歴史と、今後の方向性についてより詳細に議論する予定である。
謝辞:本研究は大村一太氏、森本元太郎博士、大野洋介博士、長谷川亜樹氏との共同研究に基づくものです。また日本IBMサービス・日立製作所・東京エレクトロンデバイスのエンジニアの皆さんには大変なご支援をいただきました。ここに感謝します。
参考文献
- Shaw, D. E., Deneroff, M. M., Dror, R. O., Kuskin, J. S., Larson, R. H., Salmon, J. K., … & Wang, S. C. (2008). Anton, a special-purpose machine for molecular dynamics simulation. Comm. ACM, 51 (7), 91-97.
- Ohmura, I., Morimoto, G., Ohno, Y., Hasegawa, A., & Taiji, M. (2014). MDGRAPE-4: a special-purpose computer system for molecular dynamics simulations. Phil. Trans. Roy. Soc. A 372 (2021), 20130387.
略歴
1964年東京都生まれ。1987年東京大学理学部物理学科卒業、1992年東京大学大学院理学系研究科物理学専攻博士課程修了(博士(理学))。同年より東京大学教養学部基礎科学科第二助手、1997年統計数理研究所助教授。2002年理化学研究所ゲノム科学総合研究センターチームリーダー、2007年プロジェクト副ディレクター、2008年基幹研究所システム計算生物学研究グループグループディレクター、2011年4月より生命システム研究センター生命モデリングコア長、2013年9月より現職。専門は計算機科学、計算生物学。
タンパク質-化合物複合体の量子化学計算-酵素反応の解明と創薬への応用に向かって-
直島 好伸
岡山理科大学自然科学研究所・解析シミュレーションネットOKAYAMA
スーパーコンピュータ「京」に代表されるコンピュータ計算能力の飛躍的な向上と優れた計算化学ソフトウェアの開発により、有機化合物のような低分子のみならず、タンパク質などの生体高分子や実験系の巨大分子の高精度計算機シミュレーションが現実のものとなっている。従来、有機合成などの研究の大半は、実験による試行錯誤を繰り返し、解決策を見出して成功へと近づく“経験重視型研究”であった。それはそれで重要なことであるが、現在のようなシミュレーション技術が発達した時代にあっては、併せて計算化学シミュレーションによる“予測先導型研究”を実験研究者自らが積極的に行うことも、自己の研究の質を高め、更なる発展を目指すために大切なことであろう [1, 2]。
最近、我々は、生体触媒の一つである酵素リパーゼを利用する光学活性化合物の不斉合成に加え、タンパク質-有機化合物複合体の全電子状態計算を自らの手で行っている。生体におけるタンパク質の酵素反応あるいはタンパク質に対する医薬品の作用などは、本質的にはタンパク質の電子レベルでの構造やタンパク質と有機化合物などの外来物質の電子レベルでの相互作用に起因している。 したがって、そういった反応や作用機能の解明にはタンパク質の電子状態を正確に知ることが重要であり、そのためのシミュレーションは電子の振る舞いを記述できる量子化学に基づいた全電子計算が極めて有用である。
本講演では、酵素リパーゼの有機合成基質に対する鏡像体選択性を予測し、且つその鏡像体認識機構を解明するために挑戦している、リパーゼ-有機合成基質複合体のABINIT-MP/BioStation [3] によるフラグメント分子軌道 (FMO) 計算、およびFMO相互作用計算に挑戦し、それに深く関わったことから図らずも参加することになった「FMO創薬コンソーシアム」(で)の活動について紹介する。FMO法はタンパク質などの巨大分子をフラグメントに分割し、部分エネルギーを集積することで系全体の高速電子状態計算を実現する方法であり [4]、 近年、製薬企業での利用が進んでいる。
【酵素リパーゼの鏡像体選択性に関するFMO相互作用計算】 有機合成で多用されているBurkholderia cepacia lipase (BCL) や Candida antarctica lipase typeB (CALB) と第1級および第2級アルコール系エステルの両鏡像体との複合体に対し、FMO2-MP2/6-31G レベルでのFMO計算を行った。鏡像体選択性が高い基質エステルの場合、優先的に変換される一方の鏡像体の全てが、それぞれのリパーゼの特定のアミノ酸残基と強く相互作用していることが判明した。選択性が低い基質では、両鏡像体ともに同じようなアミノ酸残基と相互作用している様子が認められた [5]。相互作用アミノ酸残基の特定は、実験では得られない成果である。
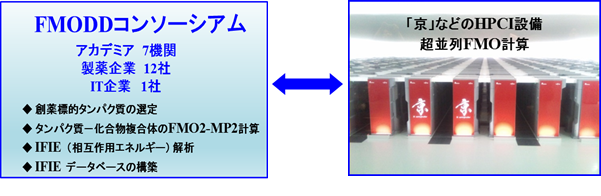
【FMO創薬コンソーシアム】 FMO Drug Design (FMODD) コンソーシアムは、FMO法によるインシリコ創薬手法を合理的で実用的な技術として発展させるために、2014年11月に設立されたもので、2015年5月現在、製薬企業12社、IT企業1社、アカデミア7機関から成る。2015年1月にはFMODDコンソーシアムキックオフミーティング、4月には同第2回全体会議が行われた。また、本コンソーシアムに関連して、2015年度のHPCIシステム利用研究課題の「京」実証利用枠における「HPCIを活用したFMO創薬プラットフォームの構築」が採択された。目下、我が国におけるFMOインシリコ創薬研究の本格的始動に向かって、創薬標的タンパク質を4つのグループに分け、スーパーマシン「京」による実質的なFMO計算を準備中である。
参考文献
- 直島好伸、“はじめての分子化学計算 実験化学者や実験生化学者がコンピュータで有機分子や生体分子をつくり計算する”、Wako Infomatic World, No. 20, 2-7 (2010).
- 直島好伸、文部科学省 次世代IT基盤構築のための研究開発 「イノベーション基盤シミュレーションソフトウェアの研究開発」 最終成果報告会 依頼講演、“量子生体分子化学計算から学んだこと”、東京大学生産技術研究所 革新的シミュレーション研究センター編、講演集、147-157 (2013).
- ABINIT-MPおよびBioStation Viewerはhttp://www.ciss.iis.u-tokyo.ac.jp からダウンロードできる。
- G. D. Fedorov, T. Nagata, and K. Kitaura, “Exploring chemistry with the fragment molecular orbital method”, Phys. Chem. Chem. Phys., 14, 7562-7577 (2012).
- Y. Yagi, T. Tanaka, A. Imagawa, Y. Moriya, Y. Mori, T. Kimura, M. Kamezawa, and Y. Naoshima, “Large-Scale Biomolecular Chemical Computations toward the Prediction of Burkholderia cepacia Lipase Enantioselectivity”, J. Adv. Simulat. Sci. Eng., Special Issue on Recent Advances in Simulation in Science and Engineering, 1, 141-160 (2014).
略歴
1976年 大阪府立大学大学院農学研究科農芸化学専攻博士課程修了 農学博士
1977年 岡山理科大学理学部助手
1978年 岡山理科大学理学部講師
1982年9月-1983年8月 米国オハイオ州立ライト大学化学科研究員
1984年 岡山理科大学理学部助教授
1992年 岡山理科大学理学部教授
1997年 岡山理科大学総合情報学部教授
2011年 岡山理科大学自然科学研究所教授(現職)
医薬品設計のための大規模分子シミュレーション
山下 雄史
東京大学先端科学技術研究センター
薬をつくるには莫大な費用と長い時間がかかる。現在、克服できていない疾病や新たに現れる病気に対応するためには、薬の開発をもっと効率的にしていく必要がある。これまでさまざまな工夫で効率化が試みられてきたが、我々がおこなっている「スーパーコンピュータを使った薬づくり」の研究も、薬づくりを効率化するためのものの1つである。我々の研究では、病気の原因となるタンパク質に強く作用する分子を探すために、大規模な計算をスーパーコンピュータでおこない、解析して薬づくりに応用することを考えている。また、そのための新しい解析手法や理論を一方で開発している。したがって、我々の研究分野の内容は、基礎的・学問的な側面から応用的・技術的な側面まで多岐に渡るものになっている。
学問的な側面から見れば、薬のデザインは「分子を認識するメカニズムを知る」ということになるだろう。多くの薬は、病気の原因となるタンパク質の機能を抑えることで、薬としての役割を果たす。もしも薬が目標のタンパク質だけでなく、いろいろなタンパク質の機能に影響を及ぼすとしたら、致命的な副作用を引き起こす可能性が高まる。したがって、目標のタンパク質だけを「認識」して攻撃できなければならない。
分子がどれくらい強く目標のタンパク質を認識できるかを表す物理量の1つが「結合自由エネルギー」である。結合自由エネルギーの値が大きい分子は、標的のタンパク質を強く認識して攻撃できるため、薬として非常に有望なものになる。我々は、この結合自由エネルギーを正確に予測することができれば、薬開発の大きな武器になると考えている。そこで、我々は高精度な結合自由エネルギー計算法を用いて、実際のがんの原因タンパク質に応用を試みている。この計算法では、1つの分子と標的タンパク質の結合自由エネルギーを評価するだけでも大きな計算資源が必要である。薬として適した分子を探すためには、多くの分子を計算して調べる必要があるので、その分さらに巨大な計算資源が必要となる。これは、従来のコンピュータでは実行不可能な計算量であった。しかし、スーパーコンピュータ「京」の登場により、ある程度、実行可能な時代に入ったと考えられる。
ここで、計算機シミュレーションの利点について強調しておきたい。第1の利点は、直接には見ることができない水中のタンパク質の様子を、高精度な顕微鏡を覗くように見ることを可能にする点である。第2の利点は、本当は起こり得ないバーチャルな状況をつくることが可能な点である。実は、このバーチャルな状況を上手く活用することで、現実の物理量をより高速に計算する工夫ができるのである。実際、我々の結合自由エネルギー計算の方法もこうしたアイデアを活用している。計算機だからこそ可能な方法を開発していくも、この分野の大きな魅力である。
本講演では、分子シミュレーションの基礎から「京」を用いておこなってきた分子シミュレーションの薬づくりへの応用研究まで、丁寧に解説していく予定である。
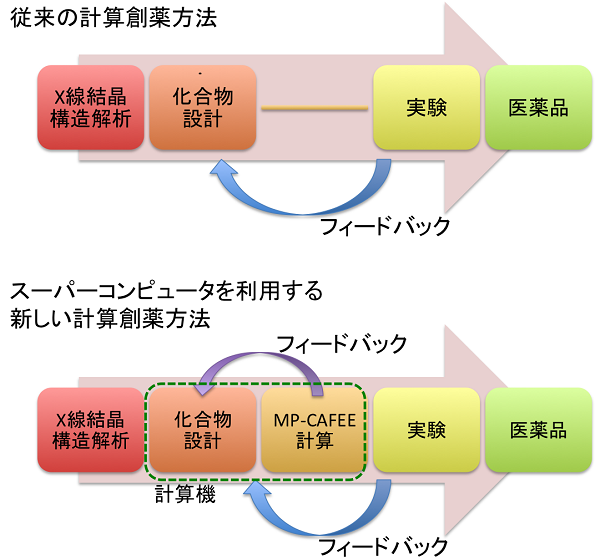
図1:スーパーコンピュータを活用する新しい計算創薬の方法
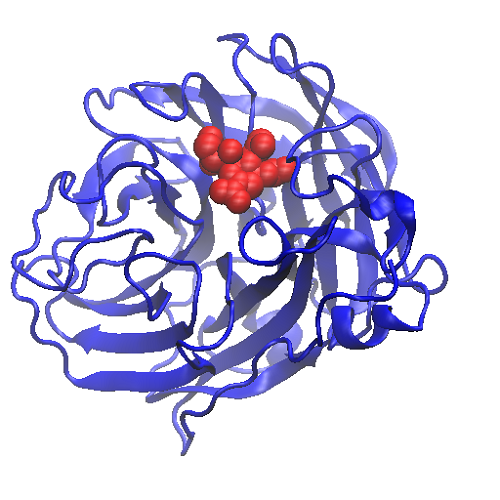
図2:標的タンパク質(青いリボン)に結合する医薬品(赤い球)
参考文献
- 山下雄史, 児玉龍彦, 月刊「化学」(化学同人), 70(2), 33-38 (2015).
- T. Yamashita et al., Chem. Pharm. Bull., 62, 661-667 (2014).
- T. Yamashita et al., Chem. Pharm. Bull., 63, 147–155 (2015).
- 山下雄史, アンサンブル (分子シミュレーション研究会会誌), (in press).
- T. Yamashita and H. Fujitani, Chem. Phys. Lett., 609, 50-53 (2014).
- T. Yamashita, JPS Conf. Proc., 5, 010003 (2015).
- T. Nakayama, E. Mizohata, T. Yamashita et al., Protein Sci., 24, 328-340 (2015).
略歴
2004年京都大学大学院理学研究科博士課程修了。東京大学大学院総合文化研究科、ユタ大学化学科、シカゴ大学化学科の博士研究員を経て、 2011年から現職。
系統樹解析と分子動力学計算に基づく[NiFeSe]ヒドロゲナーゼの分子進化の3Dシミュレーション
田村 隆
岡山大学大学院環境生命科学研究科
1. 研究の背景とねらい
Desulfovibrio属細菌に代表される硫酸還元菌(Sulfate-Reducing Bacteria)は,硫酸イオンSO42−を電子受容体とする硫酸呼吸によって旺盛に生育する偏性嫌気性菌です。SRBは,最も古くから地球に棲息していた細菌の1つと考えられており,さまざまな無機物・有機物を呼吸の基質として利用します。なかでも,水素の酸化反応を可逆的に触媒するヒドロゲナーゼ(Hase)がペリプラズム空間に複数,常備されており,水素をエネルギー源として利用します。Haseは水素の酸化還元反応を触媒する一方で,分子状酸素には極めて弱く,大気中の分子状酸素で失活する脆弱性のために,本酵素の産業利用のネックになっていました1)。
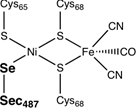
近年,石油掘削機を激しく腐食して破損させるある種のSRBから活性中心にセレノシステイン (Sec)残基を持つ[NiFeSe] 型Haseが発見され,これが2%程度の酸素存在下でも触媒能を維持したので注目を集めました。本講演では,[NiFeSe] 型Haseの分子進化を系統解析とMD計算を用いて立体構造としてシミュレーションした事例を紹介します。
2. 硫酸還元菌由来[NiFeSe]Haseの系統樹解析と大陸大移動
[NiFeSe]型HaseはD. vulgaris及び D. baculatum由来に限られていましたが,相同性検索の結果D. alaskensis, D. africanus, D. salexigens, D. pigerなど近縁のSRBゲノムにもホモログ遺伝子がコードされていました。ゲノムのデータベース上では終始コドンとされているUGA が実はSecをコードするセレン含有酵素であると示唆されました。セレンタンパク質の発現には,オパールコドン(UGA)をSecとして翻訳するためにはセレノソームと呼ばれるSelA, SelB, SelC, SelDの遺伝子群が必要とされます2)。これらのSRBゲノムには,必要とされる遺伝子群がコードされており,未同定ながら[NiFeSe] Haseが複数,存在することを強く示唆しています。
[NiFeSe] Haseは一体どこからやってきたのか。という素朴な疑問と興味から,系統樹解析を行ないました。最尤法など複数の系統樹解析した結果,[NiFeSe] Haseは同一のグループに収束したので,[NiFeSe]型Haseは共通祖先から派生したことが伺えます。バクテリアは高等生物とは異なり,遺伝子の水平伝播やある特定の時期に爆発的に分子進化が加速するために,タンパク質の系統から種の系統を辿るのは,困難なのですが,複数の遺伝子の共進化関係を辿ることで,細菌ゲノムの系統関係を考察するアプローチも提案されています。セレノソームを構成する遺伝子群は,クラスターではなくそれぞれのゲノム上のあちこちに散在していますが,それらの系統樹も[NiFeSe]Haseと相似形を示したことから, Haseの分子進化は,種の系統と共に分化してきたと示唆されました。つぎに,この系統樹をGUIDANCEアルゴリズムでアライメント検定した後にANCESCONで過去の配列を再現すると12種類の[NiFeSe]HaseをもつSRBは陸生由来(うち2株は,ヒト腸内からの病原菌)と海生由来に分岐していました。海生にも絶対嫌気環境である海底熱水噴出口と微好気環境である海底堆積物に棲み分けた系統に分かれていました。また系統樹的に近縁な[NiFeSe]酵素を持つ分離株の地理的分布から,現存する株の共通祖先がパンゲア旧大陸の住人であったこと,地球環境の変動に伴う大陸大移動によって世界に分布したことも考察されました。
3. 分子動力学計算が再現するタンパク質の分子進化
[NiFeSe]型Haseの結晶構造解析(2wpn.pdb)から,本酵素は活性中心と分子の表面をつなぐ大きなGas Cavityを持つことが明らかにされています。タンパク質の内部のNi-Fe活性中心と表面をつなぐ巨大なトンネルは,水素分子の取込みには有利ですが,酸素分子のアクセスも同時に容易になるというデメリットもあり, Cavity形成の分子進化にはトレードオフがあったと考えられます。
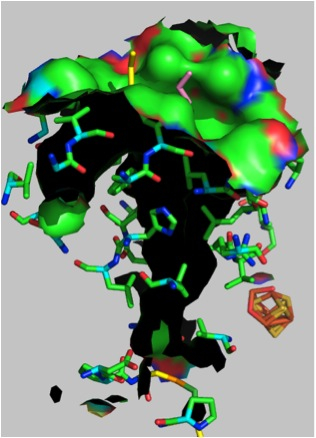
[NiFeSe]Haseのガスキャビティ
結晶構造2wpnを鋳型として,祖先型配列および現存配列の立体構造を構築しました。ホモロジーモデリングはかなり粗いモデル化なので,クラスター計算機による分子動力学(MD)計算を12Åの深さの周期境界水モデルにおいて,11ナノ秒,300K,1気圧下の条件で最適化を図りました。これを23個の祖先・現存の[NiFeSe]Haseについて行う膨大な計算と構造解析(3年かかりました)により,最初に活性中心にSec残基を獲得した共通祖先酵素から分岐した分子進化とガスキャビティの変遷を再現しました。まず共通祖先にはガスキャビティが形成されておらず,絶対嫌気的環境が確保された油田や海底熱水噴出口から分離された株の[NiFeSe]Haseに至る分子進化においては,共通祖先から徐々にGas Cavityが発達する経緯が再現されました。しかし,酸素発生型の光合成生物と共存した環境下において酸素暴露リスクの高い堆積層に棲息しているD. postgatei, D. autotrophicumが持つ[NiFeSe]Hase への分子進化では,Cavityが未発達の閉じた立体構造がシミュレーションによって描かれました。これらの株が棲息する環境では,昼間は光合成型生物が活発に酸素を発生させるので極めて好気的な環境が形成されています。そこでペリプラズムに局在する[NiFeSe]Haseも酸素暴露のリスクが高いためにこのような穴を発達させなかったと考えられます。分子進化を駆動する非同義置換(δN)のアミノ酸残基がCavity形成に果たす役割について検討した結果,全体の5%を占めるδNのアミノ酸残基が,残り95%の高度に保存された領域のコンフォメーションを支配しているという新しい発想を提唱するに至りました。
参考文献
- P. M. Vignais, B. Billoud, Occurrence, classification, and biological function of hydrogenases: An overview. Chem Rev 107, 4206-4272 (2007).
- A. Böck et al., Selenocysteine: the 21st amino acid. Mol Microbiol 5, 515-520 (1991).
略歴
平成 5年 京都大学大学院博士後期課程修了 (農学博士)
同年 岡山大学農学部農芸化学科 助手
平成12年 岡山大学大学院自然科学研究科 助教授
平成25年 岡山大学大学院環境生命化学研究科 教授
数学と大規模シミュレーションから読み解くがんの自然史
波江野 洋
九州大学理学研究院 生物科学部門 数理生物学研究室
元々正常であった細胞が制御不能になり、異常増殖を始める。それらの細胞は他組織に浸潤し体中に広がっていく。このようながんの基本的な進展には、突然変異が大きく関わっている。毎日億というオーダーの細胞が30億塩基対という長さの塩基配列を複製する。複製の際のエラーに対する監視機構、修復機構は存在するものの、60年、70年と正常に機能し続ける事は不可能である。突然変異の起こる場所によっては細胞に影響を与えないが、細胞分裂の制御などに関わる重要な遺伝子の配列に変異が起こった場合、細胞の制御機構が1つ崩れる事になる。このような突然変異が細胞に蓄積していく事によってがん細胞が生じる。
組織の中で突然変異が蓄積していく過程を数理モデルで表し、その原理・原則を見いだそうとする研究が2000年代から急速に発展してきた。数理モデルを用いる利点としては、(i) 現実には50年から60年かかるがん発症の過程をコンピュータ上で数秒から数時間という短い時間で表現することが出来るということ、(ii) 細胞増殖率、突然変異率などの条件を変化させて、その影響を簡単に調べられること、(iii) 現実のデータをうまく表す数理モデルを構築できれば、その後のがんの動態を予測することが出来ること、などが挙げられる。
私はこれまでに数理解析とコンピュータシミュレーションを用いて、がん細胞が発生・進展する過程、転移を起こす過程、投薬の中で薬剤耐性を獲得する過程を研究してきた。未だに克服が難しいがんという病気に対して、発生からヒトを死に至らしめるまでの過程(がんの自然史)を明らかにすることで、がん進展の様々な時期における防衛策を提示することを目指している。本講演ではこれまでの研究の中から、臨床データを用いた膵臓癌における転移発生過程とBRCA関連癌の進展に関する数理解析研究を紹介する[1,2]。
膵臓癌は年々増加傾向にあり、現在は臓器別がん死亡率で肺がん、胃がん、大腸がん、肝臓がんに次いで第5位である。膵臓癌はその高い浸潤能、転移能のため固形がん最大の難治がんであり、5年生存率が5%程度と予後が極めて不良である。膵臓癌の唯一の根治的治療法は外科的切除であるが、切除をしても潜在的な遠隔転移を有している事が多く再発することが多い。近年の研究では、p16, p53, smad4遺伝子が遠隔転移能の獲得に関連するとされており、外科的切除時に変異している遺伝子数と生存予後との相関が報告されている。
今回、これらの遺伝子変異を有するがん細胞、有さないがん細胞を仮定し、各細胞の増殖、死亡、変異、転移イベントを想定して出生死亡過程によるシミュレーションを行い、膵臓癌の臨床像におけるがん進展の再現を試みる。また、患者さんが診断された後、外科的切除や放射線治療、化学療法による治療の介入にて、最も生存予後を良くする方法を考察する[1]。
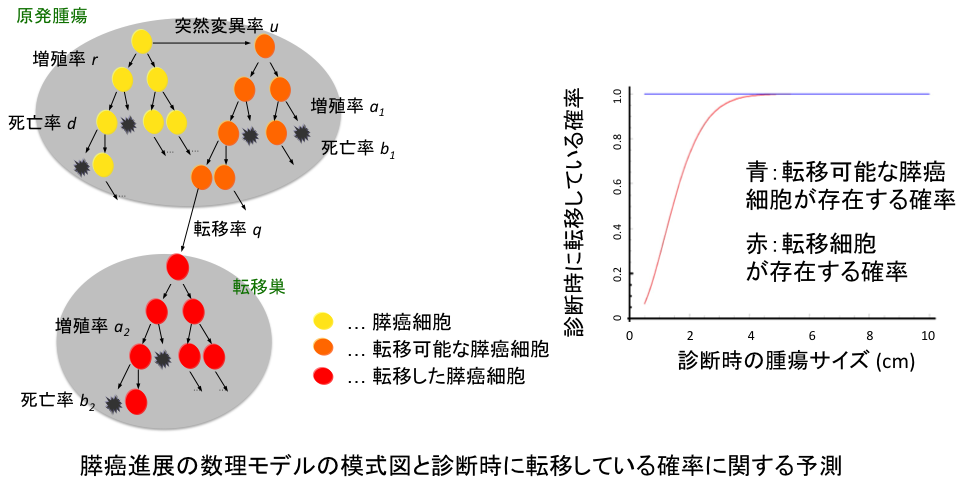
BRCAの不活性化は卵巣癌や乳癌でよく見られ、DNA修復に異常をもたらすことによって発がんの起点になることが知られている。BRCAが不活性化しているがん細胞ではDNA修復機構が十分に機能しておらず、様々な突然変異が起こり易い。そのことを利用して、DNAに傷をつけることによってがん細胞を選択的に殺すプラチナ製剤やPARP阻害薬が治療の選択肢として考えられるが、プラチナ製剤の投与後にがん細胞が薬剤耐性を獲得することも報告されている。本研究では正常細胞が突然変異の蓄積によってBRCAを欠失しがん細胞として異常増殖する過程を数理解析し、BRCA関連癌が発症する過程でプラチナ製剤に対する薬剤耐性を獲得する仕組みを明らかにし、プラチナ製剤を投与した方が良い条件を提示する[2]。
参考文献
- Haeno et al. Computational modeling of pancreatic cancer reveals kinetics of metastasis suggesting optimum treatment strategies. Cell 2012
- Yamamoto et al. Evolution of pre-existing versus acquired resistance to platinum drugs and PARP inhibitors in BRCA-associated cancers. PLoS One 2014
略歴
2010年九州大学大学院生物科学専攻において学位取得。その後、ハーバード大学生物統計学部門・ダナファーバーがん研究所計算生物学部門においてMichor博士の研究室で博士研究員に従事する。2013年4月から現職。多種多様ながんについて、がんの一生を数理モデルで理解することを目指している。
国際連携による癌の全ゲノムシークエンスとスーパーコンピュータ
中川 英刀
理化学研究所 統合生命医科学研究センター
がんは正常細胞のゲノム(約30億=3GbのDNA配列)に様々な異常が蓄積し、正常な分子経路が破綻した結果、無秩序な細胞増殖や転移をきたす「ゲノムの疾患」である。今日、これらがんのゲノム異常を標的とした様々な分子標的治療薬が開発され、がんの治療成績の向上に寄与している。乳がんのHER2遺伝子増幅を標的としたハーセプチンが成功例であり、EGFRを標的とした薬が開発されてEGFR変異のある症例が適応となる一方で、その下流のKRAS遺伝子に変異があるものは適応外となり、ゲノム情報をバイオマーカーとしたがん治療の個別化が進みつつある。また、BRCA1/2遺伝子の変異のある遺伝性乳がん患者やその家族には、予防的切除などの様々な措置もとられようとしており、ゲノム情報を指針したがん予防医療も広まりつつある。
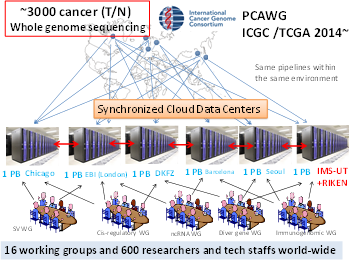
超大量のDNA配列を決定する次世代シーケンス技術(Next Generation Sequencing: NGS)とゲノム情報解析技術の爆発的な進歩に伴い、個人のヒトゲノムの配列約30億塩基=3Gbを短期間で決定し、ほぼすべての変異/多型の検出が可能な全ゲノムシークエンス解析(WGS)が比較的安価で迅速に行うことが可能になってきている。現在の最先端のNGSシステムでは、年間18000例のWGSが1000ドル/例のコストで可能である (3Gx30x18000=1500 Tera塩基/年)。しかしながら、これだけの配列情報を扱うための計算リソースと効率のいい解析方法および人材が不足しているのが、問題である。
「ゲノムの疾患」であるがんに対して、発生機序の解明と理解、新規治療法や個別化医療の発展を目的として、国際連携にてがんゲノムをシークエンス解析し、そのデータを共有・公開する国際がんゲノムコンソーシアム(International Cancer Genome Consortium:ICGC)が2008年に創立され、現在は16か国+EUが参画し、80もの様々はがんのゲノムシークエンスプロジェクトが遂行されている。日本では理化学研究所と国立がんセンターが肝臓がんを担当し、270例のWGS解析および250例の全エクソンシークエンス解析(たんぱく質をコードする1-2%の領域)が、東大医科研のスパコン「Shirokane」を駆使して完了した。これまで、ICGC全体で12800例のがんのゲノムデータが登録・公開されており、様々ながん研究で活用されている。そして2014年より、米国のがんゲノムプロジェクトTCGAとICGCと共同で、PCAWG (PanCancer Whole Genome Sequencing Project)が開始され、TCGA/ICGA内での2500-3000例の様々ながん種のWGS(全体で約600 Tera塩基)を、6つのゲノム解析に特化した世界中のスパコンを同調させながら、600人もの研究者が共同で解析を行っている。理研は、この全データの約10%(肝がん270例)に貢献し、東大医科研スパコンと共同で6つのゲノムデータ解析拠点の1つを運用してこの国際連携ゲノムプロジェクトに深く貢献してきている。
今後、がんゲノムに限らず、ゲノムシークエンスデータの産出は急激に増加することは間違いなく、計算リソースや人材の確保、および大量のゲノムデータから、いかに生物学的な「解釈」を導き出すが、最大の問題点である。
参考文献
- Nakagawa H, Wardell CP, Furuta M, Taniguchi H, and Fujimoto A. Cancer whole genome sequencing: present and future. Oncogene (2015) doi: 10.1038/onc.2015.90.
- Fujimoto A, Furuta M, Shiraishi Y, et al. Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat Commun 6: 6120 (2015)
- Fujimoto A, Totoki Y, et al. Whole genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet 44: 760-764 (2012)
- The International Cancer Genome Consortium. International network of cancer genome projects. Nature 464:993-998 (2010)
略歴
平成 3年(1991) 3月 大阪大学医学部医学科卒業
平成 3年(1991) 4月 大阪大学医学部第2外科 臨床研修医
平成 4年(1992) 6月 大阪大学医学部付属病院ICU レジデント
平成 5年(1993) 6月 国立大阪病院 外科レジデント
平成 8年(1996) 4月 大阪大学大学院医学系博士課程
平成 8年(1996) 6月 大阪大学医学部第2外科 医員
平成11年(1999) 9月 米国 The Ohio State Univeristy ポスドク
平成15年(2003) 4月 東京大学医科学研究所 ヒトゲノム解析センター 助手
平成19年(2007)11月 同 准教授
平成20年(2008) 2月 理化学研究所 ゲノム医科学研究センター チームリーダー
平成25年(2013) 4月 理化学研究所 統合生命医科学研究センター チームリーダー







