
要旨
- スーパーコンピュータの贈り物 -過去、現在、未来-
- 生体分子のための第3世代密度汎関数法の開発
- スパコンと数学で迫るがんの個性
- 電子状態計算から生体分子物性・創薬へ
- スーパーコンピュータ「京」による創薬イノベーション
スーパーコンピュータの贈り物 -過去、現在、未来-
江口 至洋
理化学研究所 HPCI計算生命科学推進プログラム
1.過去
ここでは「スーパーコンピュータ」を「その時代時代の高性能な電子計算機」としてとらえます。それでは、スーパーコンピュータはいつ誕生したのでしょう。正確なことは不明ですが、有名な高性能電子計算機にENIACがあります。ENIACは1946年の2月に公開された電子計算機で、現在みなさんが使用されているスマホやパソコンの能力には全く及びません。しかし、当時としては極めて高性能な電子計算機、スーパーコンピュータです。公開現場に立ち会ったニューヨーク・タイムズの記者は、「多くの計算が、まばたきをする間もなく終わってしまう」と驚きを持って記事にしています。その頃すでにフォン・ノイマンらは新しい「ノイマン型電子計算機」の開発を始めます。開発にあたって、フォン・ノイマンはスーパーコンピュータでなければ解けない問題は何だろうと考え、電子計算機の開発チームとともに気象予測のチームを同時に立ち上げています。その成果は1952年に実ります。このインパクトは大きく、1954年から1959年にはスウェーデン(気象・水理局)、アメリカ(気象局)、日本(気象庁)がスーパーコンピュータを導入し、数値予報研究を始めます。日本で数値予報が現場で活用され始めたのは1980年代のことですが、当時の研究者を奮い立たせる力をフォン・ノイマンは与えてのです。同じ頃、ノーベル物理学賞を受賞したフェルミも「スーパーコンピュータでなければ解けない問題」としてバネで繋がれた多くの質点の動きの研究を始めます。バネの力がフックの法則に従うと、その問題は「紙と鉛筆」で解くことができます。フェルミらはバネの力が非線形である問題を解こうとしました。その計算結果は、当初の予想から全くかけ離れたものでした。この研究はしばらく表舞台から去りますが、1965年にはソリトンという新しい波の発見につながります。
ここで重要なことは、計算機科学の進歩とともにあらゆる学問分野で計算科学が20世紀後半に生まれてきたということです。その橋渡しをスーパーコンピュータはしてきました。「スーパーコンピュータの贈り物」は次のように整理することができます。
(1) 実験を助け、補完します
(2) 実験できないものを、「実験」します
(3) 人間の予測能力を向上させます
(4) 人間の思考能力を超えます(?)
(5) 新しい発見をもたらします
(6) 学問を結び付け、学際性を強めます
2.現在
1946年のENIACから2011年のスーパーコンピュータ「京」までの65年間で計算性能は10兆倍以上も進歩したと言われています。現在「京」を用いた計算が民間企業や大学等研究機関を含め多くの研究者によって進められています。それらは正に、現在における「スーパーコンピュータの贈り物」となっており、そのうち戦略分野での成果を以下に例示的に示します。
分野1 予測する生命科学・医療および創薬基盤(理化学研究所)
分子レベルの挙動を取り入れた心臓シミュレータや血栓形成シミュレータの開発を行い、病態の解明や医療への応用を進めています。
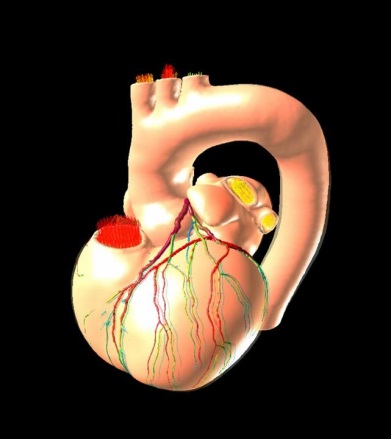
東京大学 久田、杉浦、鷲尾、岡田研究室
協力:富士通株式会社
分野2 新物質・エネルギー創成(計算物質科学イニシアティブ)
「京」により、第一原理分子動力学計算に基づく化学反応過程の解明が可能となり、電池の電極とその周りの電解質との反応の計算が可能となりました。
分野3 防災・減災に資する地球変動予測(独立行政法人海洋研究開発機構)
従来は積乱雲を詳細に表現することは分解能の限界から困難でしたが、「京」により分解能を高めることができ、個々の積乱雲から全球規模の積乱雲群との相互の関係をより正確に調べることが可能になりました。
分野4 次世代ものづくり(東京大学生産技術研究所)
既存の風洞実験では走行中の自動車が遭遇する様々なリスク(追い越し、追い抜き、突風、急なハンドル操作)の予測を行うことは困難でしたが、「京」での計算機シミュレーションにより可能となりました。
分野5 物質と宇宙の起源と構造(計算科学連携拠点)
従来、超新星爆発のシミュレーションや、星形成、恒星進化は空間分解能、時間方向とも3次元計算には能力が不足していました。「京」によって、超新星爆発の仕組みのシミュレーションが可能になりました。
3.将来
20世紀前半までの科学は主に実験と理論の両輪で進められてきました。20世紀後半には計算科学が生まれ、その存在感を徐々に増してきました。21世紀には実験科学と理論科学、そして計算科学が相互補完を進め、科学は急速に進歩してきています。スーパーコンピュータはその進歩の名補佐役として、みなさんに多くの新しい贈り物を今後も届けていきます。
生体分子のための第3世代密度汎関数法の開発
佐藤 文俊
東京大学 生産技術研究所
レアアース、ナノ構造物質、タンパク質は、わずかなエネルギーで反応が進む分子である。いずれも密に詰まった複雑な電子状態を利用しているが、そのような電子状態を作り出す要因は様々である。レアアースはレアアース元素の電子構造そのものが、ナノ構造物質は幾何学的に特徴ある構造がそれを担っている。一方、地球上に豊富に存在する元素でできているタンパク質は、巨大な構造を持つことで特異な電子構造を作り出している (図1)。したがって、タンパク質の反応を定量的に解析するためにはタンパク質全体を丸ごと取り扱う必要がある。
数千~数万原子からなるタンパク質の電子状態を計算する方法は、現在、大きく分けて2種類ある。1つは、QM/MM法 (2013年ノーベル化学賞)、divide-and-conquer法、フラグメント分子軌道法といった、領域分割法である。もう1つは、分子サイズの4乗 (計算方法によってはそれ以上) に比例する計算量を主に数学的な技術を用いて減少させる、スケーリング法である。当グループでは、タンパク質の電子状態を本質的に理解するために [1]、後者の立場で標準の正準分子軌道法を研究開発している [2]。特に、機能性の高い金属を持つタンパク質に興味があり、計算方法には密度汎関数法を採用している [3]。
新たな計算方法やアルゴリズムの開発には、発展し続ける計算機システムの構成を十分に考慮する必要がある。今や、TOP 500に入るスパコンは、「京」同様、分散メモリ型超並列計算機である。これらのマシンを使う意義があるためには、99.9999…%といった並列化率と、高い単体性能の両者を満たさなければならない。相当に高いハードルである。計算機システムが計算方法を制限する事態になってはいないだろうか。さらに、ポスト「京」はこれに加えて加速器が付く可能性が高い。果たして、使いこなすことができるのであろうか。
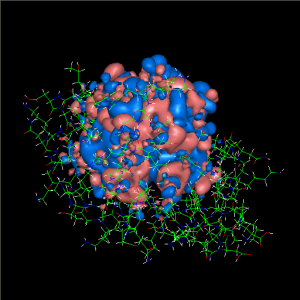
図1: シトクロムcの分子軌道
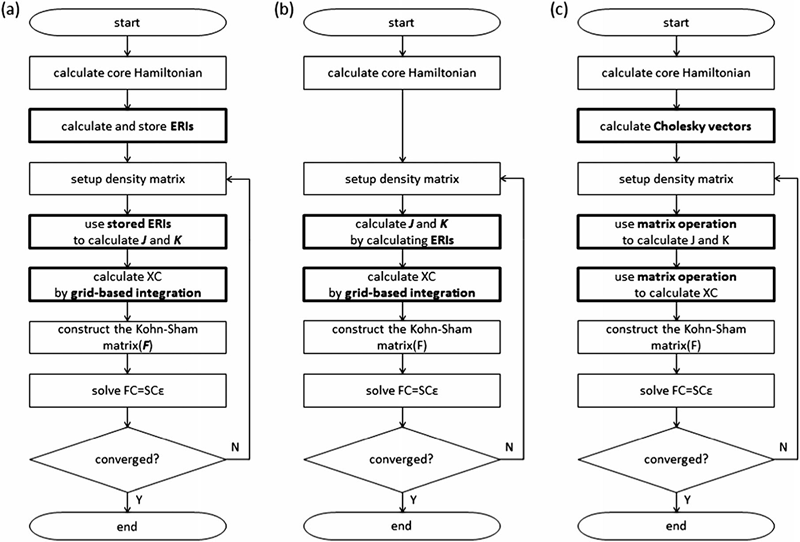
図2: (a)ファイル法、(b)ダイレクト法 (現行法)、(c)第3世代法
当グループは、これまで現行法のダイレクト法による正準分子軌道計算の超並列化に注力してきた [3]。しかし、これでは4か所の異なるアルゴリズムによる計算律速ルーチンをそれぞれチューニングしなくてはならず、計算機システムの開発との間でイタチごっことなっていた。そこで最近、密度汎関数法の純交換相関ポテンシャル項を見積もるためのグリッドフリー法の改良に成功したのを機に、正準分子軌道計算に基づく第3世代密度汎関数法を提案した [4]。本方法は、コレスキー分解を利用し、4中心分子積分を線形演算可能な行列として保持する。SCF繰り返し計算前に一度だけこの計算を行い、SCF計算中は行列演算のみで正準分子軌道計算が達成できる (図2)。行列演算には、巨大行列を各計算ノードに分散保持し、ScaLAPACKなどの線形演算ライブラリを用いればよい。線形演算ライブラリはTOP 500の評価のために、どのような計算機システムでも必ず最大限努力により最適化チューニングされる。したがって、第3世代法ではSCF計算前の分子積分とコレスキー分解計算のチューニングに注力すればよく、「京」のみならず原理的にはポスト「京」でも極めて効率良く大規模分子の電子状態計算が可能となる。恐らく、ポスト「京」では、Born-Oppenheimer全電子分子動力学計算を駆使した、タンパク質の本格的な化学反応シミュレーションが実行できる。基礎研究のみならず、新規酵素設計や薬剤最適化などの応用シミュレーションに威力を発揮するだろう。
なお、以上のように、シミュレーションプログラムは開発だけでなく、頻繁に発展を遂げないと直ぐに古くなってしまう。数十万ラインからなるプログラムの開発・アップデート・保守・管理にはソフトウェア工学が有効であるが、これをそのまま適用すると弊害が多い。そこで、シミュレーションソフトウェアに適したソフトウェア工学を提案した [5]。シミュレーションソフトウェアの開発に興味のある方は、是非ご一読を。
参考文献- K. Chiba, T. Hirano, F. Sato, M. Okamoto, IJQC, 2013, 113, 2345–2354.
- 上村典子, 佐藤文俊, 恒川直樹, 西村康幸, 平野敏行, 吉廣保, 甘利真司, 加藤昭史, 小林将人, 田中成典, 中野達也, 福澤薫, 望月祐志, “プログラムで実践する 生体分子量子化学計算 -ProteinDF/ABINIT-MPの基礎と応用”, 佐藤文俊, 中野達也, 望月祐志編, 森北出版, (2008).
- 佐藤文俊, 恒川直樹, 吉廣保, 平野敏行, 井原直樹, 柏木浩, ”タンパク質密度汎関数法”, 柏木浩監修,森北出版, (2008).
- T. Hirano, F. Sato, PCCP, 2014. DOI: 10.1039/C3CP55514C.
- 佐藤文俊, 高橋英男, 居駒幹夫, “ソフトウェア開発入門: シミュレーションソフト設計理論からプロジェクト管理まで”, 佐藤文俊, 加藤千幸編, 東大出版, (2014).
スパコンと数学で迫るがんの個性
井元 清哉
東京大学医科学研究所 ヒトゲノム解析センター
がんは、親から受け継いだ遺伝的要因(ゲノム)、腫瘍細胞に蓄積した遺伝子変異(がんゲノム)、環境要因によるゲノムの修飾(エピゲノム)、これらの違いや異常が、正常な細胞の営みを司っている遺伝子ネットワークやシグナル伝達・代謝などのパスウェイに入り込み、システム異常を起こした時空間で進化するヘテロな細胞集団である。そして、血管内皮細胞や免疫炎症細胞などの正常細胞を操り、抗がん剤に対して耐性を獲得していく。ゲノム変異が大きく異なっている複数の原発が進化することも報告されている。こうした複雑さを背景にして、がんは抗がん剤などに対する薬剤感受性や予後の良・不良等、様々な個性を持つ。そして、そのシステム異常の中心で遺伝子の発現を調整しているメカニズムが遺伝子ネットワークであり、がんの個性の一つの捉え方である。
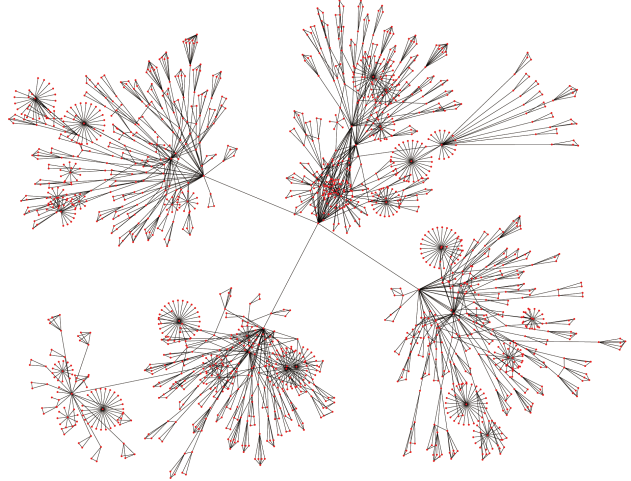
図1:遺伝子ネットワークの例
我々は、多様ながんの個性を遺伝子ネットワークとして抽出する方法として NetworkProfiler という方法を開発した。NetorkProfilerは、がん細胞から計測したメッセンジャーRNAの発現データから遺伝子ネットワークを推定する数学的な方法で推定する方法である。数百というがん細胞のメッセンジャーRNAの発現データをNetworkProfilerにより統合解析することにより、それぞれのがん細胞が有する予後の良し悪しや、ある抗がん剤に対する耐性感受性などその特徴を規定する遺伝子ネットワークを見いだすことができる。もし、ある抗がん剤が効かないがん細胞らに共通に活性化されている遺伝子ネットワークが見いだされれば、その遺伝子ネットワークを不活化するような化合物を加えた後に抗がん剤を投与することにより、抗がん剤が効かなかったがん細胞に効果が見られることが期待される。
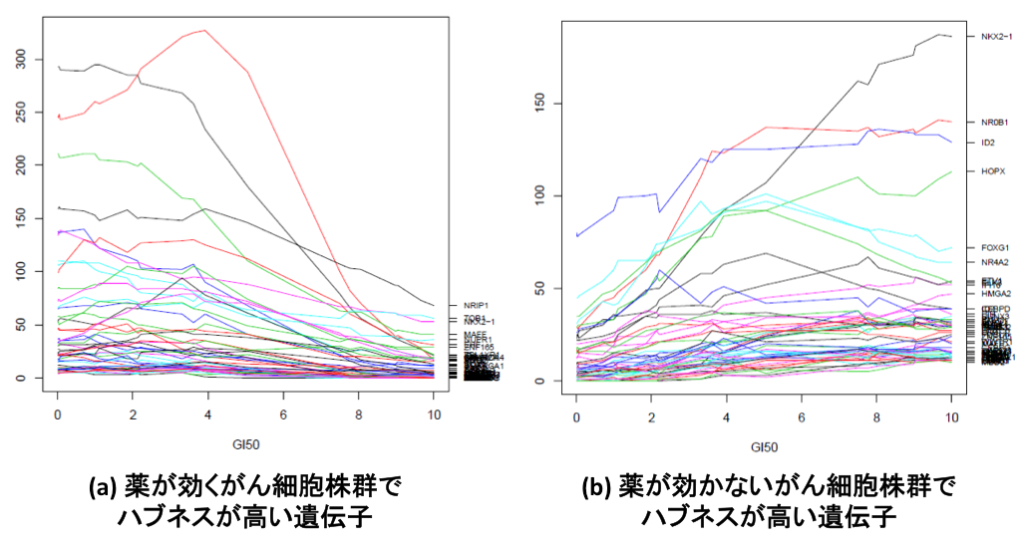
図2:抗がん剤エルロチニブが効くがん細胞株と効かないがん細胞株の遺伝子ネットワーク比較
英国サンガーセンターが公開している Sanger Genomics of Drug Sensitivity of Cancerの遺伝子発現データをNetworkProfilerにより解析し、抗がん剤に対するがんの多様な抵抗性をネットワークとして抽出した。このデータセットでは、13,435遺伝子のメッセンジャーRNAの発現データが728のがん細胞株において計測されており、142種類の抗がん剤や医薬品候補化合物についてのIC50の値も合わせて計測されている。我々は、このうち101種の薬剤について解析を行った。この解析では、101種の薬剤それぞれの効果を規定している遺伝子ネットワークを全ての細胞株それぞれで推定するため、合計7万近い遺伝子ネットワークを推定する必要がある。我々は、この計算にK Computerを利用し、8000コア約3日間の計算により約7万個の遺伝子ネットワークの計算に成功した。
T. Shimamura, S. Imoto, Y. Shimada, Y. Hosono, A. Niida, M. Nagasaki, R. Yamaguchi, T. Takahashi, S. Miyano (2011) A novel network profiling analysis reveals system changes in epithelial-mesenchymal transition, PLoS ONE, 6(6), e20804.2.
電子状態計算から生体分子物性・創薬へ
田中 成典
神戸大学大学院 システム情報学研究科
生命機能を理論的に記述・理解し、その成果を医療やエネルギー問題の解決などに役立てるための一つの方法論として、生体を構成する原子・分子のレベルから第一原理的(非経験的)にモデリングしていくアプローチが考えられる。昨今のハイパフォーマンス・コンピューティング(HPC)技術の進歩により、生命系の基本分子であるタンパク質や核酸の全電子計算も比較的容易になっており、電子状態計算に立脚した定量的に信頼できる分子間相互作用解析・反応解析が可能となり、既に疾患メカニズムの解明やドラッグデザインに生かされている。我々が研究開発を進めてきたフラグメント分子軌道(Fragment Molecular Orbital; FMO)法による解析事例も、インフルエンザウイルスの変異予測や種間感染機序の解明、核内受容体のリガンド結合予測、ホタルなどの生物発光現象の解明など多岐に亘っている[1]。本講演では、こうした状況を踏まえ、生命機能のボトムアップ的な記述を目指す計算機シミュレーションにおける現在の問題点のいくつかを取り上げ、また、今後の方向性について論じたい。
ひとくちに生命機能の分子レベルからの記述と言っても、例えばタンパク質の電子状態の解析と個体レベルで見られる現象の描写の間には大きな階層的なギャップがある。細胞レベルの記述でさえも、生体分子個々の取り扱いのレベルから見ればかなり遠いという印象があるかもしれない。これに関しては、生体反応ネットワークの総体的モデリングを指向した、いわゆるシステム生物学的なアプローチが有効な橋渡しをしてくれる場合が多い。我々のグループでも最近、光化学系IIを例にとって、タンパク質レベルでのミクロな反応を光合成系全体のマクロな応答に定量的に関係づける数学的手法の開発を進めている[2]。こういったアプローチの成功は、ボトムアップの立場からは、酵素反応や光反応などの個々のタンパク質レベルでの振舞いを、その周囲の環境効果を適切に取り込みながら可能な限り定量的に正確に(実験の助けも借りて)記述することの重要性を再認識させてくれる。「細胞内混み合い」の問題は昨今のホットな話題の一つであるが、生体分子認識だけでなく、例えば生体分子系における電子移動やエネルギー移動が周囲の環境条件に大きく支配されることは古くから知られてきた。FMO法などに基づく第一原理的な解析手法をいかに有効に活用して反応速度定数等の現実的かつ正確な評価を行うかといった重要な課題が残されている[3,4]。さらに、生化学反応の多くは、水溶液中で行われるダイナミカルなプロセスであり、第一原理シミュレーションの立場からは、分子動力学(MD)による構造変化(ゆらぎ)の追跡とそれに伴う電子状態とくにフロンティア軌道の変動の記述が同時に(かつ自己無撞着に)行われなければならない。FMO法の枠組[1]では、このことはFMO-MD法とFMO-LCMO(Linear Combination of Molecular Orbitals)法を効率的に併用することにあたり、これはHPC技術の助けなしには達成が困難なタスクとなる。また、以下の創薬応用の問題においても現れるが、水素結合、プロトン化状態、また、周囲のイオンの存在[5]なども構造変化や反応にとって本質的な影響を与える。
実用的な観点から言うと、生体分子系に対する第一原理電子状態計算の成果をインシリコ(in silico)創薬に活用することは社会的な貢献・インパクトが大きいと考えられる。FMO法もその開発の歴史を通じて、さまざまな創薬応用がなされ、他の手法では得られない知見や有用性を提供してきた[1]。例えば、フラグメント間相互作用エネルギー(Inter-Fragment Interaction Energy; IFIE)を用いたリガンド分子とアミノ酸残基間の網羅的な相互作用解析は、タンパク質-リガンド複合系における結合エネルギーの詳細分析を可能にする点で、クラスター分析や定量的構造活性相関の手法などと組み合わされてドラッグデザインの新たな地平を切り拓いた。最近では、より詳細なフラグメント分割を可能にするFMO4法や相互作用エネルギー分割の手法などを用いることで、いわゆるFragment-Based Drug Design(FBDD)の理論的基盤を提供している。また、リガンドドッキングを行う際の力場やスコア関数の改良にFMO計算の結果を用いる試みもなされつつある。従来の分子力学計算で用いられる力場ではタンパク質-リガンド相互作用に伴う分極や電荷移動の効果を取り込むことが困難な場合が多いが、複合体構造に対する電子状態計算の結果から原子電荷パラメターの値を決め直す[6]ことでドッキングの精度を著しく改善することが期待されている。
第一原理的なインシリコ創薬の展開にも多くの課題が残されている。量子力学的なシミュレーションは対応する古典力学的な計算と比べてはるかに多くの計算量を要求し、そのことは計算結果の信頼性が最初に採用した初期構造に依存する度合いが高いことを意味する。量子力学的な構造最適化の高コスト性から、従来は信頼できる生体分子構造が他から提供されることを「待つ」という状況が続いていたが、今後は、X線結晶構造解析の高分解能化そのものにFMO計算を活用するといったプランが提案されている。1Å以下の分解能を有するタンパク質の構造決定においては、プロトン化状態やプロトン位置の精密決定の問題が顕在化するが、量子力学計算はこれに対しても解決を与えうる。また、リガンド結合の自由エネルギー変化を見積もる上では脱水和の記述が重要となるが、FMO計算の枠組の中で溶媒効果、水素結合、遮蔽効果、エントロピー変化などをどのように扱うかについても今後の展開が待たれる。
参考文献- S. Tanaka, Y. Mochizuki, Y. Komeiji, Y. Okiyama and K. Fukuzawa, Phys. Chem. Chem. Phys. 16 (2014) 10310.
- T. Matsuoka, S. Tanaka and K. Ebina, BioSystems 117 (2014) 15.
- S. Tanaka and E.B. Starikov, Phys. Rev. E 81 (2010) 027101.
- Y. Suzuki and S. Tanaka, Phys. Rev. E 86 (2012) 021914.
- M. Nakano, H. Tateishi-Karimata, S. Tanaka and N. Sugimoto, J. Phys. Chem. B 118 (2014) 379.
- Y. Okiyama et al, Chem. Phys. Lett. 467 (2009) 417.
スーパーコンピュータ「京」による創薬イノベーション
奥野 恭史
京都大学大学院 医学研究科
医薬品の開発は、10年以上の長い年月と、500億円以上の巨額の費用がかかると言われている。またこの十数年、製薬業界では、世界的規模で、新薬の承認数が横ばい状態(20品目程度/年)であるのに対し、研究開発費が増え続けるという深刻な問題に直面している。この医薬品開発の高コスト化は、製薬会社に対する直接の経済的負担になっているばかりでなく、超少子高齢化社会を迎える日本の医療費に対しても間接的影響を与えている。つまり、開発費用が高くなればなるほど薬の価格も高くなり、それにともなって医療費も高くなってしまうのである。このことから、医薬品開発の効率化をはかり、開発コストを下げることは、製薬業界のみならず、日本の医療費問題にとっても重要な課題となっている。このような背景から、近年、医薬品開発を効率化する有力なアプローチとして、計算機シミュレーションによる創薬に大きな期待が寄せられている。しかしながら、現状の創薬計算技術には未だ残された課題が多く、新薬創出を強力に加速する技術革新が待ち望まれてきた。
このような中、我々は製薬企業、IT企業、アカデミアの連携で産学コンソーシアムを設立し、スーパーコンピュータ「京」を用いた革新的な創薬計算技術の開発に挑戦してきた。コンソーシアム名 ”K supercomputer based drug discovery (KBDD) コンソーシアム” 製薬企業18社、IT企業2社、アカデミア5機関から成る大所帯である。KBDDコンソーシアムでは「京」の圧倒的なマシンパワーを用いることで、創薬計算分野が抱える以下の2つの根本的課題に挑戦している。また、これらの課題を克服するための計算が実現できたとしても、結果を得るために日常業務を逸脱した複雑な操作を必要としたり、数か月もの計算時間を要したりするようでは、産業利用では何の役にも立たない。このような観点から、創薬現場で「京」を実践的に利用できる計算フローの構築も我々の目標とした。
【課題1】医薬品候補化合物の探索では、膨大な化合物候補(10の60乗以上の化合物数)と多数の創薬標的タンパク質候補との莫大な組合せ数の相互作用評価を行うことが理論上必要であるが、これまでの創薬計算技術では特定の標的タンパク質に対して数百万の化合物のバーチャルスクリーニングしかなされていない。
【課題2】医薬品候補化合物の探索と最適化での現状の創薬計算技術の予測精度は、平均5%程度であることから、タンパク質と化合物との結合親和性を頑強かつ正確に予測できる計算技術の確立が急務である。
我々はこれらの課題を解決するために、「課題1」に対しては、独自に開発する高速かつ高精度な化合物探索計算法「Chemical Genomics-based Virtual Screening 法(CGBVS法)」(文献1)を「京」に実装し、化合物とタンパク質の大規模相互作用空間の超高速探索に挑戦した。CGBVS法は大規模相互作用データを機械学習することでバーチャルスクリーニングを可能にしたものであり、「ビッグデータ創薬」の先駆的取り組みとして位置付けられる。
一方、「課題2」に対しては、化合物とタンパク質との結合自由エネルギーの高精度な推定が可能である「Massively Parallel Computation of Absolute binding Free Energy(MP-CAFEE法)」(文献2)を「京」に実装・チューニングし、化合物とタンパク質との結合自由エネルギーの精密な予測に挑戦した。MP-CAFEE法はアンサンブル型の分子動力学(MD)計算を通じて、タンパク質と化合物の結合自由エネルギーを算出するものであり、「シミュレーション創薬」の本格的取り組みと位置付けられる。
本講演では、KBDDコンソーシアムで実施してきた「京」による「ビッグデータ創薬」と「シミュレーション創薬」の研究開発成果について紹介する。

- Yabuuchi H. et.al. Mol. Syst. Biol., 7, 472 (2011)
- Fujitani H. et.al. Phys. Rev. E, 79, 021914 (2009)







