
課題4 大規模生命データ解析
平成25年度~27年度における実施計画
研究開発課題の概要
本研究開発課題は、ゲノムを基軸とした大規模生命データ解析により生命プログラムとその多様性を理解することを目標としている。
細胞から地球規模のシステムを構成している共通の基本情報がゲノムであり、ゲノムには生命システムを構成する部品の設計図とそれらの部品を組み立てて機能を作り出すおおよそのプログラムが記述されている。ヒトゲノムの場合、平均でDNA 300文字につき1か所の頻度で個人ごとに少しずつ異なっており、また同じがん腫でもゲノム異常は多様である。この多様性が、薬の効き方や、治療後の予後に影響していると考えられている。
最先端のシークエンサーの登場により、ゲノムのDNA配列だけでなく、DNAの修飾状態、DNAのコピー数、機能性RNAを含む転写産物を超高速・低コストで網羅的に解析できるようになった。このため、メタゲノム解析や脊椎動物10Kプロジェクトなど地球規模でゲノム解析が進行し、人類はパーソナルゲノム時代を迎えようとしている。加えて、様々の最先端計測技術が生命システムデータの精緻化・大規模化を加速している。しかし、その背後にある生命システムの設計原理は、最も明らかになっている遺伝継承の原理を除き、転写制御ネットワークやシグナル伝達ネットワークなどについては多くの生物学的知見が蓄積されてはいるが、あまり明らかではない。そのため、物理モデルを高度に並列化するというアプローチではなく、大規模データ解析という方法論が有効である。
こうした背景の基、本研究開発課題の目標を達成するために、京コンピュータに最適化した最先端・大規模シークエンスデータ解析基盤を整備した上で、生命プログラムの複雑性・多様性や進化をゲノムによって理解する研究と同時に、ゲノムを基軸とした生体分子ネットワーク解析研究を行う。大規模生命データ解析の基礎となるデータは公開されているものを用いる他、データ解析の結果出てくる予測や正当性の検証などについては、国際がんゲノムコンソーシアムをはじめとする大規模ゲノムプロジェクト、がんやメタボリック症候群などの病気に関する大小の病態解析プロジェクトと連携して実施する。それにより、これまで手の付けようの無かった規模のデータ解析を大規模計算で実施し、薬効・副作用予測、毒性の原因の推定、オーダーメイド投薬、予後予測などへの応用に貢献することを目指す。
研究開発課題の内容
超並列シークエンサー技術で生産されるゲノムを基軸とした大規模生命データ解析を京コンピュータを用いて実施し、がんのシステム異常及びその個性と脂肪細胞の機能分化のプログラムの理解を目標とし、次の3項目を狙った研究を展開する。
(1)パーソナルゲノムと生物多様性の理解の深化
(2)細胞やがんの個性を見いだし、それらの背後にある分子ネットワークを描出する
(3)病態を御すための方法論、及び医療介入の予測法の構築
国際がんゲノムコンソーシアムは2万5千人のがん検体からゲノム異常のカタログを完成させる。しかし、がんのゲノム情報だけで個々人のがんの個性を全遺伝子規模のシステムとして細やかに描きだすことには限界がある。そのため、個々人のがんの病態に密接に関係する遺伝子ネットワークを大規模スケール(多人数)で全遺伝子規模で解析することが一つの挑戦的課題となる。また、長い時間を経て変貌したヘテロながんゲノム空間やヒトゲノムが体内微生物ゲノムと織りなす共生空間と病態の関係を解読することは、数千CPU程度のコンピュータを使った配列解析やモデリングでは非現実的な時間を要する未踏の世界となっている。さらに、細胞の機能分化の仕組みの解明に向けて分子生物学的方法論に基づき膨大な研究が展開されているが、それによって描き出せる生命プログラムは小規模・局所的である。そこで、環境要因などの刺激後、新たな機能をもった細胞へと変化するプロセスを網羅的に遺伝子ネットワークの変化として描き出し機能形成の秘密を解き明かすことも大きな挑戦である。本研究課題では、京コンピュータを活用し、本課題及びグランドチャレンジプログラム「次世代生命体統合シミュレーションソフトウェアの研究開発」で開発した高並列アプリケーションを大規模に実行し、これらの挑戦的課題をブレイクスルーする。
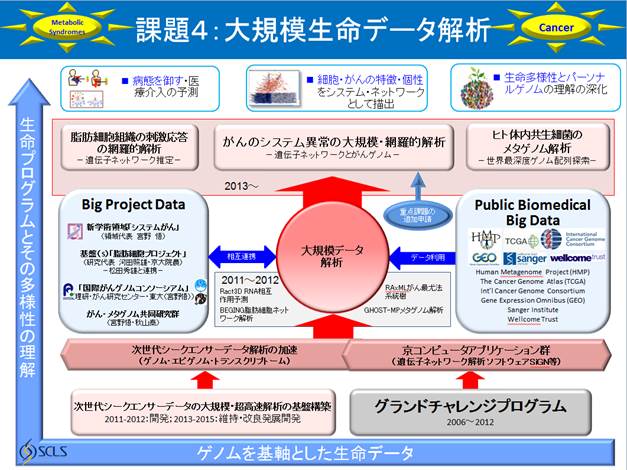
このために、第1期(平成23年度~24年度)と第2期(平成25年度~27年度)に分けて、以下のサブテーマに取り組む(年次計画参照)。
第1期(平成23年度~24年度)
1) 次世代シークエンサーデータ解析のための情報処理システムの開発(秋山 泰・東京工業大学)
京コンピュータに最適化した次世代シークエンサーデータ処理・解析ソフトウェアを整備する。このために平成23年度~24年度に研究費をこの開発に集中する。実装レベルで高速化のための様々な工夫(ファイルストライピング、OpenMPによるスレッド並列性のチューニング、等)により、京に最適の独自の計算手法を開発し、京による大規模データ処理の道を拓く。応用としては、既存環境でも時間さえ掛ければ扱い得る単純な配列マッピング処理ではなく、高感度でなければ発見できない未知遺伝子群または変異配列に対する高度なマッピングおよび相同性解析や、計算量の大きいde novoアセンブリ処理等の機能を京で実現し、今後重要となるがんゲノムやメタゲノムなどの難度の高いゲノム解析の支援を行う。まず、パラメータを調整することで高速・高感度に大規模な配列解析を迅速に行うことを可能にするGHOST-MPを開発し、ヒトメタゲノム解析を行う。性能としては5億リード/時を達成する。
本課題の目的は、従来の近似的なマッピング処理では発見できないような、新規遺伝子および変異配列を扱える高感度な配列解析を、従来の数千から数万倍高速に実現することであり、がんゲノムやメタゲノム解析では、従来不可能であった新規の発見を支援することが可能となる。成果のデータベース化は当課題の直接の目的ではないが、HPCI計画で現在議論されているアーカイブ機能が実現された場合には、解析成果をデータベースとして公開することも視野に入れる。
2)RNA相互作用予測技術の開発と転写物の網羅的情報解析(浅井 潔・産業技術総合研究所)
ゲノム多様性と生命システムデータの多様性の間には複雑な生体内分子ネットワークが存在している。生命プログラムを解明するために、この生体内分子ネットワークを捉えるモデルを生命システムデータから構築し、ゲノム多様性と合わせてデータを統合化して理解することが必要である。このためにはノンコーディングRNA中心として、その相互作用の予測が重要な技術であることに間違いはない。そこで、転写物RNA間相互作用の予測システムのパイプラインを開発する。このパイプラインは、配列情報解析に基づく3段階のステップと、立体構造解析するステップからなる。配列情報解析の第1ステップは、RNA配列の2次構造的解析を行い、各RNA配列に2次構造的アノテーション(予測2次構造、部分配列に対するアクセサビリティなど)を施すものでありCentroidFold (Hamada M et al. Prediction of RNA secondary structure using generalized centroid estimators. Bioinformatics 25(4):65-473, 2009)や Raccess (Kiryu H et al. A detailed investigation of accessibilities around target sites of siRNAs and miRNAs. Bioinformatics 27(13):1788-1797, 2011)等を用いる。第2ステップは、RNA配列の相補性と2次構造アノテーションを用いて相互作用する配列組候補を抽出するものであり、SlideSortやLASTを活用する。第3ステップは、配列組候補における2次構造的相互作用解析により、相互作用する部位とその強度を予測するものであり、RactIP (Kato Y et al. RactIP: fast and accurate prediction of RNA-RNA interaction using integer programming. Bioinformatics 26(18):i460-i466, 2010)を京に実装して、大規模な計算を実施する。立体構造解析では、フラグメントアセンブリと分子シミュレーションによってより精密なRNA間相互作用を解析する。また、相互作用の部位だけでなく強度を比較するためのアルゴリズムとソフトウェアを開発する。立体構造解析のフラグメントアセンブリでは、単体のRNAの2次構造を活用して立体構造予測する開発済みソフトウェアRASSIEを用い、より柔軟な2次構造予測情報に対応しできる手法に拡張する。立体構造解析の分子シミュレーションでは、修飾塩基を含む2次構造パラメータの抽出を行うとともに、2次構造予測情報を活用した相互作用予測のための手法を開発する。
3)大規模な生体分子ネットワークの解析技術の開発(松田秀雄・大阪大学)
グランドチャレンジプロジェクトにおいて、宮野悟(東大)は大規模遺伝子ネットワーク推定プログラムのパッケージSiGNを開発しており、京を活用すれば、10万ノードからなる生体分子ネットワークの構造推定、数千~数万遺伝子からなる遺伝子制御ネットワークの高精度推定、さらに遺伝子数を30程度に絞りこめば最適ベイジアンネットワークを計算することができる。このソフトウェアにより、DNAチップで解析した遺伝子発現データに基づき、グローバルなネットワークから局所的なネットワークまで、バイアスのかからない探索が実現できることになる。しかし、このように推定したネットワークを解釈する際には、ハブ遺伝子などに着目し、抗がん剤耐性やがん遺伝子の変異情報などの生物学的・医学的知見をアドホックに重ねながら解析を進めるしかないというボトルネックがある。一方で、こうした生物学的・医学的探求の焦点はますます多様化している。そのため、これまでの研究で蓄積された生物学的・医学的な知見を取りこみ、解析の焦点を明確にして、限られたデータからより高精度にネットワークを推定することにより、このボトルネックを解消することが不可欠になっている。また、次世代シークエンサーによるRNA-Seq定量データは未知の機能性RNAをネットワーク解析に取り込むことができるという利点がある一方、DNAチップデータのようにノードにすべきmRNAやmicroRNAが定まっておらず、定量化も含め、大規模ネットワーク解析はそこから開始しなければならない。そこで本研究では、これらの問題を解決できる大規模な生体分子ネットワークの解析のための研究開発を実施する。まず、複数の生物学的知見から構成したシードネットワークを与えることで、その間で生じる生体分子ネットワークの構造変化の解析が行えるソフトウェアBENIGNの開発を行う。これより、細胞分化で、幹細胞と分化後のネットワーク群を入力して、細胞分化過程を推定することを試みる。次に、この開発をベースにして、次世代シークエンサーのデータ解析等から得られる膨大な量の生体分子データをもとに、大規模な生体分子ネットワーク解析を行えるように拡張する。そして、多様に変化する条件ごとの生体分子ネットワーク解析から、生命プログラム全体の構造の推定するソフトウェアへ発展させ、生命プログラムおよびその多様性を、生体分子ネットワークの観点から解明することを目指す。また、細胞分化など、関連する研究者と必要な協議等を行う。
4)メタゲノム・比較ゲノム解析研究 (五條堀孝・国立遺伝学研究所)
生命プログラムの複雑性・多様性や進化は、原始生命から連綿と継承されてきた遺伝情報担体である「ゲノム」に刻まれている。飛躍的に発展するシークエンサーから得られる膨大なデータを解析する上で問題となるのは、これらの情報をいかに取り出すか、ということである。比較ゲノムによる手法は、このようなデータを用いてメタゲノムを含めた生物多様性を理解するために磨き上げられた最も有効な手法であり、系統樹推定はその一部である。系統樹推定は、ゲノムから得られる塩基配列情報に多重整列処理が施された複数の配列から系統樹推定を行うものである。その一つである最尤法は、近年の計算機性能の向上に伴って生命科学分野の研究において頻繁に利用されており、評価が安定している手法である。最適な樹形を推定するためには候補系統樹を網羅的に探索する必要があるが、候補樹形の数は、配列データ数をNとすると、O(2NN!) のオーダーで爆発的に増大するため (種数N=32に対しても2192となる(2180=(100京)3)、一定数以上の配列に対しては計算資源上の制約から、探索空間を絞る必要がある。本課題は、系統樹推定を解析の基盤として、京コンピュータの計算パワーを最大限引き出し、大規模ゲノム解析に適用することで、生命科学にゲノムの観点から新たな光を当てようとするものである。系統樹推定には様々な応用が考えられるが、本課題では、①日本人の人類集団における進化的位置、②感染症ウイルスの進化的解析、および③がん細胞の系統的関係の解析の3つを目標とする。初めのテーマでは、ヒトゲノムの多型を集団毎に詳細化することで、ゲノムコンテクストが集団毎にどのように異なり、変化しているかを明らかにする。2番目のテーマでは、ウイルスの網羅的系統樹を構築し、病原性等に関わる因子が、宿主側のゲノムコンテクストとどのように関わっているかを解析し、ウイルスの感染リスクとの関係について評価する。3番目のテーマでは、がんの病巣がステージ進行に従って患者の体内でどのように転移・進行していくのかをがん細胞の系譜図を作成する事によって明らかにする。
以上の準備と研究に基づき、第2期では、RNAの構造予測については浅井潔(東大)、がんの細胞集団の進化については五條堀孝(国立遺伝学研究所)、データ同化技術については樋口知之(統計数理研究所)、遺伝統計学については角田達彦(理化学研究所)、並びに、がんや脂肪細胞の実験系・臨床系研究者に業務協力者として連携し、以下の研究開発を実施する。
第2期(平成25年度~27年度)
5)大規模データ解析によるがんのシステム異常の網羅的解析とその応用(宮野 悟・東京大学)
がんは、親から受け継いだ遺伝的要因(ゲノム)、腫瘍細胞に蓄積した遺伝子変異(がんゲノム)、環境要因によるゲノムの修飾(エピゲノム)、これらの違いや異常が、正常な細胞の営みを司っている遺伝子ネットワークやシグナル伝達・代謝などのパスウェイに入り込み、システム異常を起こした時空間で進化するヘテロな細胞集団である。そして、血管内皮細胞や免疫炎症細胞などの正常細胞を操り、抗がん剤に対して耐性を獲得していく。ゲノム変異が大きく異なっている複数の原発が進化することも報告されている。こうした複雑さを背景にして、がんは抗がん剤などに対する薬剤感受性や予後の良・不良等、様々な個性を持つ。そして、そのシステム異常の中心で遺伝子の発現を調整しているメカニズムが遺伝子ネットワークであり、がんの個性の一つの捉え方である。
本研究では、多数のがんサンプルデータを用いて大規模・網羅的に遺伝子ネットワークを中心に解析し、そのような多様な個性を生み出すがんのシステム異常の実態をシステムとして解明することを目的とする。これにより、がんの生命プログラム及びその多様性の理解を深め、薬効・副作用予測、毒性の原因の推定、オーダーメイド投薬、予後予測などへの応用に貢献することを目指す。国際がんゲノムコンソーシアムは、50種のがんについて全部で25,000人のサンプルをシークエンスし、がんのゲノム異常のカタログをやがて完成させるが、これだけでがんの多様な個性をシステムとして細やかに捉えることには限界があり、個々人のがんの病態に密接に関係する遺伝子ネットワークを網羅的に解析することが、がんの複雑さと個性を理解するためのチャレンジとなっている。しかし、ゲノムワイドな遺伝子ネットワーク解析だけでも大きな計算コストを要し、その網羅的解析は京コンピュータの稼働以前では夢物語であった。そこで、グランドチャレンジプログラムで開発したアプリケーションSiGNなどを利用・発展させ、京コンピュータの資源を十分に用いることで、以下の研究を実施する。
700以上のがん細胞株の遺伝子発現プロファイルデータと100以上の薬剤に対するがん細胞株の薬剤感受性(IC50スコア)データを、複数のゲノムワイドな遺伝子ネットワーク推定法で解析し、薬剤感受性とがん細胞株との関係をシステムの違いとして調べる。また、数万の臨床検体の遺伝子発現プロファイルデータ及び関連するオミクスデータと臨床データを用いた大規模・網羅的遺伝子ネットワーク解析を行う。これらの大規模データ解析によってがんの病態に関する有用な知見が得られることが期待されるが、業務協力者との連携により、知見またはその発展形の検証を様々な観点から実施する。 以上の研究を遂行する中で必要となる新たな大規模生命データ解析の方式の研究を併せて実施する。さらに、計算資源が十分に利用できれば、以下のテーマを追求する。
(i) 肺腺がんのシステム異常をゲノム、遺伝子ネットワークの観点から大規模・網羅的に解析し、予後の予測が困難とされるステージI肺腺がんの予後を支配するシステム異常の本態に迫る。
(ii) がん組織体・転移組織の全体ゲノムの進化と遺伝子発現状態を3次元エクソーム解析とトランスクリプトーム解析によりとらえ、システム異常の変化と転移・再発等の悪性度の関係に迫る。
また、大規模生命データ解析の課題全体の統括を行い、「大規模生命データ解析」に関わる研究者との必要な協議等、並びに連携先等との調整を行う。
6)大規模生体分子ネットワーク解析による脂肪細胞組織の刺激応答の網羅的解析とその
応用(松田秀雄・大阪大学)
本研究では、大規模生体分子ネットワーク解析による脂肪細胞組織の刺激応答の網羅的解析とその応用のための研究開発を実施する。マウスの脂肪細胞の組織的な変化を、in vivoの時系列発現プロファイルでネットワーク解析することが特色である。培養細胞ではなく、マウスの臓器からRNAサンプルを直接取っており、臓器の細胞の組織的な変化がとらえられる。マウス白色脂肪細胞中のベージュ細胞(特定の刺激を受けて褐色化する細胞組織)は、エネルギー代謝の活発なヒト成人の褐色脂肪細胞に近いことが報告されている(Wu J, et al. Cell. 2012 Jul 20;150(2):366-76)。この細胞の機能分化について、従来は、細胞の表現型の変化と、個別のマーカー遺伝子の発現変動のみに注目していたが、脂肪細胞の中で起こっている「動的な変動の全体像」の解明を目指し、京の計算資源を用いて、大規模・網羅的探索を行う。この研究は、より一般的には、組織細胞の分化転換(transdifferentiation)という最近注目されているトピックにつながるものである。また、iPS細胞などの遺伝子導入による細胞の分化誘導は、がん化などの問題があるため、元々その個体に備わっている細胞を、ある種の刺激により別の細胞に分化転換をはかるのは大きな意義がある。その解析に京が貢献できれば大規模生命データ解析において大きな意義がある。将来的には、組織性幹細胞の分化誘導の制御に結び付けることを目指す。
特に、平成25年度は、種々の脂肪細胞組織が持つエネルギー貯蔵と高い熱産生能という相反する多面性を、刺激応答に対する脂肪細胞内部の生体分子の経時的変化のデータから、平成24年度までに開発した大規模生体分子ネットワーク解析のソフトウェアBENIGNを用いて、大規模かつ網羅的に生体分子ネットワークを解析することで、脂肪細胞が状態を変化させエネルギー消費に向けて働く機構と生活習慣病の改善との関係を探る。
7)次世代シーケンサデータ解析のための情報処理システムの開発(秋山 泰・東京工業大学)
本研究では、平成24年度までの研究開発を継続・発展させ、次世代シーケンサから産出される大量のゲノム配列情報の超高速解析を実現するための研究開発を実施する。
特に、平成25年度は、リード配列の相同性解析のための並列ソフトウェア(GHOST-MP)のコード最適化を継続して行い京での実行効率をさらに高める。またヒト体内細菌の大規模なメタゲノム解析を中心として、80,000ノード級の計算をともなう超大規模研究を実施する。既に平成24年度までに京の全系を用いた測定を一度行っているが、その後に計算機能の拡張やコード最適化を実施したので、平成25年度は80,000ノード級での性能測定を複数回実施する。また単なる性能測定ではなく、実際にヒト体内共生細菌(当面は口腔内細菌を対象)に関する大規模なメタゲノム解析を実施し、実用面における実証的な評価を得る。また、I/O負荷を相対的に軽減することを目的として、少数のノードのみがI/Oを担当し、Tofuネットワークを用いてデータのブロードキャストによりI/O衝突を減らす新手法について様々な条件下での性能評価を行い、方式として優れていればGHOST-MPの基本機能として組み込みを実施する。さらに、パイプラインへのジョブの投入および実行状態モニタなどを簡便に行うためのインタフェースの第一版の開発を終了し、京との遠隔接続の可能性および通信性能などを調査する。

具体的な成果目標
- 個々人のがんの分子病態システムや薬の作用点や副作用の原因パスウェイを明らかにすることが可能になり、また脂肪細胞などの正常な細胞分化が環境要因などの刺激により動作するプログラムの解明に貢献し、生活習慣病の改善や予防、がんという極めて複雑なシステム異常を背景とする病気への挑戦の道を拓く。
- 生体内分子、化合物、環境因子などが織りなす病態・生命プログラム及びその多様性を捉える技術を開発・応用することで、薬効・副作用予測、毒性の原因の推定、薬剤耐性遺伝子や創薬ターゲット探索などにブレイクスルーを喚起する。
- 個別化医療の基礎となる診断技術、オーダーメイド投薬、予後予測などの技術を開発する。







