
Findings
Pioneering the Future of Computational Life Science toward Understanding and Prediction of Complex Life Phenomena
Our Mission : BioSupercomputing Newsletter Vol.5 SPECIAL INTERVIEW
Development of R&D projects : BioSupercomputing Newsletter Vol.6 SPECIAL INTERVIEW
Program Director Toshio Yanagida
Deputy Program Director Akinori Kidera
Deputy Program Director Yukihiro Eguchi
Annual Report(Fiscal Year 2014)
- Theme 1 Simulations of biomolecules in cellular environments
- Theme 2 Simulation applicable to drug design
- Theme 3 Hierarchical integrated simulation for predictive medicine
- Theme 4 Large-scale analysis of life data
Simulations of biomolecules in cellular environments (Theme 1, Yuji Sugita, GL)
In this project, we are performing multi-scale simulations to study how biomolecules are working in the cellular environments. The project contains two subprojects: (i) the effect of macromolecular crowding on enzyme reactions in EGF signal pathway and (ii) multi-scale modeling of nucleosomes and chromatin. The following is the research summary of the two subprojects, which was done in the fiscal year 2014.
(1) The effect of macromolecular crowding on enzyme reactions in EGF signal pathway.
In the cytoplasm of a cell, a huge number of proteins, RNAs, and metabolites exist at high concentration. This macromolecular crowding environment has been considered as one of the key features in cellular environments and is expected to affect on protein stability, functions, and protein-protein interactions, and so on. Using K computer, we have simulated bacterial cytoplasmic systems based on the all-atom molecular dynamics (MD) simulation method. To perform large-scale MD simulations, we have utilized highly parallelizaed MD software (GENESIS: Generalized-Ensemble Simulation System), which has been developed in RIKEN AICS. In 2013, we investigated macromolecular translational and rotational diffusion in the crowded environments and compared them with coarse-grained Brownian dynamics (BD) simulations with hydrodynamic interactions. In 2014, we examined protein stability, protein-metabolite interaction, and protein-protein interaction in the cellular environments. According to statistical mechanics theory, the native globular structures are stabilized due to the excluded volume effect in the crowded environments. Recent theoretical and experimental studies have suggested that it is not always true. In our simulations, most of proteins and RNA are stabilized in the cellular environments rather than in dilute solution. However, PGK, which has two rigid domains, showed an opposite tendency mainly by electrostatic interactions. We utilized this information in the single-particle simulations on EGF signal pathway and the hybrid QM/MM free-energy calculations of enzymatic reactions in the crowded environments.
(2) Multi-scale modeling of nucleosomes and chromatin.
A nucleosome is composed of DNA wounding around histone protein cores and is a basic unit of DNA packing in eukaryotic cells. It. In most textbooks of molecular and cellular biology, hierarchical DNA structures from nucleosomes to chromosomes are described with their cartoon figures. However, there are arguments on the existences of the 30nm fibers of chromatin structures. The information about active and non-active conformations of chromatin is also limited. In our approach, we are employing multi-scale MD simulations in combined with structural information, namely, X-ray crystallography, electron micrograph, and small-angle X-ray scattering (SAXS), etc. In this year, we combined the all-atom and coarse-grained MD simulations with SAXS data and developed the MD/SAXS simulation method. This allows us to predict structures that correspond to SAXS profiles of nucleosomes in solution. We are also carrying out large-scale free-energy calculations on a single nucleosome with different histone proteins. This calculation is based on the all-atom models with explicit solvent and requires a large number of CPUs for performing hundreds of MD simulations in parallel on K computer. All the information from the multi-scale simulations and experiments has been combined to establish nucleosome structures with histone variants in solution, tri- and multi-nucleosome conformations in the cellular environments.
Simulation applicable to drug design(Theme 2, Hideaki Fujitani, GL)
In spite of the continuous progress of technologies, it is still difficult to develop a new drug and therefore more efficient drug design methods are required. In the conventional in silico drug design method, a crystal structure of the target protein is ususally used to design compounds, a few of which are selected and assayed for binding free energy. If a good candidate is found, it will be approved as a drug after careful crinical tests. However, it is quite difficult to find a good candidate on the first try and we need to design and assay several compounds with the experimental feedback iteratively. Typically, many iterations are required to find a good drug candidate, and therefore the drug development is costly and time-consuming.
In this project, we proposed a new supercomputer-assisted drug design method and examined its feasibility. [1] (See. Fig. 1.) This method involves the MP-CAFEE calculation procedure before the experimental assay. Because the MP-CAFEE calculation can predict the binding free energies for the designed molecules accurately, it is expected to accelerate the drug development procedure by decreasing the number of experimental trials.
However, the MP-CAFEE method requires huge computational cost: To predict the binding free energy for one drug candidate, the MP-CAFEE method carried out 768 molecular dynamics (MD) simulations. All the molecules are explicitly modeled at the atomic level in these simulations, while the water molecules are usually neglected in the conventional drug design programs. More importantly, the dynamical effect on the binding affinity is naturally taken into account in the MP-CAFEE method, while it is neglected in the conventional drug design programs. Therefore, while the MP-CAFEE method can predict the binding free energy accurately, it requires huge computational cost. Furthermore, in the drug design procedure, it is often necessary to predict the binding free energies for hundreds of drug candidates. In this project, we demonstrated that the extraordinary computing power of K computer enables the new drug design procedure.
In examining the free energy calculation methods, the importance of force field becomes highlighted. Among several force fields, we found the FUJI force field always provide the best values. The backbone torsion parameters of FUJI force field were determined to reproduce with LCCSD(T0)-level ab initio molecular orbital calculation results so that it can accurately describe the protein motion. We also applied this force field to MD simulations of the antigen-antibody complex system and analyzed the mutation effects on the antigen-antibody interactions. [2]
Reference
[1] T. Yamashita et al., Chem. Pharm. Bull. 63, 147–155 (2015)
[2] T. Nakayama, E. Mizohata, T. Yamashita et al.. Protein Sci. 24 328-340 (2015)
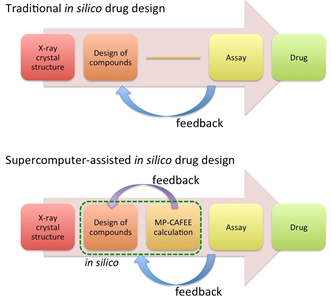
FIG. 1: Supercomputer-assisted drug design procedure
Hierarchical integrated simulation for predictive medicine (Theme 3, Shu Takagi, GL)
For the modeling of Pakinson’s disease symptom, the detail analysis was conducted by Doya’s group to reproduce the spike signals due to STN-GPe-GPi interactions in basal ganglia. They succeeded finding the parameter area to reproduce the difference of Parkinsonian state and normal state which are observed in the experiment of Dopamine-depleted monkeys. Based on this finding, they also developed the model of thalamus-cortex coupling. Fig.1 illustrates the simulation results with this model. Through the coupling of thalamus with motor cortex, the propagation of unstable wave are projected into roughly two different regions in the cortex.
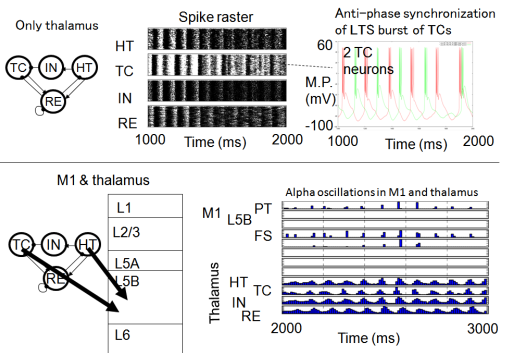
Fig. 1 Modeling and Simulations of Thalamus and Motor Cortex Coupling
This thalamus and Motor Cortex model was unified with basal ganglia model using the software called MUSIC as shown in Fig.2. One of the important achievements of this year is this unification. The different functions of different parts in brain were successfully coupled through this process and the next stage which is coupling with musculoskeletal system has been started.
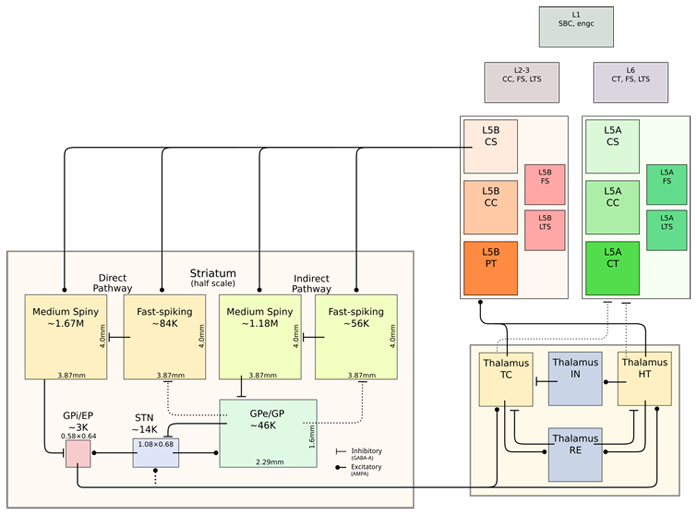
Fig. 2 Basal Ganglia-Thalamus-Motor Cortex Coupling using MUSIC
In Takagi’s group, toward the large scale parallel computing, the scalability of the software for multiple muscles system was examined. In their model, each muscle is assigned to each CPU so that the shared memory algorithm is available for finite element simulations. This year, several matrix solvers was tested for the OPEN MP simulation and Super LU for K-computer was chosen.
Nakamura’s group has been developing the model toward the entire body simulation. This year, nervous-musculoskeletal system was further developed and the detail coupling model along the spinal code was developed. One of the simulated results is shown in Fig.3
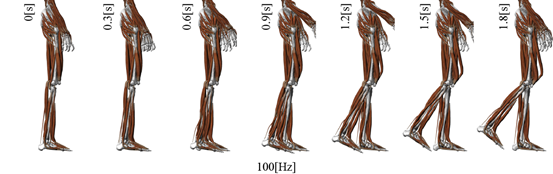
Fig.3 Musculoskeletal simulation caused by 100 spike signal imput through a-motor neuron
For circulation system, Hisada’s group has been developing the multiscale-multiphysics heart simulator: UT-Heart. Last year they developed the 3 stage hierarchical modeling from sarcomere dynamics through muscle cell to the entire heart. This year, this model has been further developed and a new sarcomere model proposed by Dr. Lorenzo Marcucci was introduced. Compared with the previous standard model, this model has a much wider valley of potential of mean force. This change of the potential curve brought the quantitatively more realistic model for ATP consumption by entire heart activity. UT-Heart has been also used to understand the remodeling of heart muscle fiber structure. Through the simulations, preferential orientation of the muscle fibers has been analyzed and the results are discussed through the comparison with actual anatomical observation. In addition to these fundamental studies, UT-Heart has been started to be used in clinical use for the after-treatment prediction of heart support net etc.
For thrombosis simulator, the large scale simulation of actual flow chamber experiment scale has been conducted. The snapshot of the simulation is shown in Fig.4. The results support the experimental results obtained by Goto’s group. The modeling of drag efficacy on different types of antiplatelet medicines ( P2Y12 Inhibitor, GPIIb/IIIa Inhibitor etc.) has been continuously studied through the comparison with the flow chamber experiment designed for the current purpose.
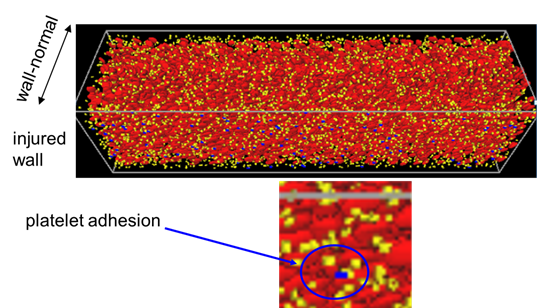
Fig. 4 Large Scale Simulation of the Platelet Adhesion on the vWF Immersed Wall (Simulation Domain: 2048x512x512, 2,048nodes(16,384 cores) on K-computer)
Large-scale analysis of life data (Theme 4, Satoru Miyano, GL)
Our biomedical targets are cancer, obesity, and metagenome. The group consists of three PI’s, Satoru Miyano (University of Tokyo), Hideo Matsuda (Osaka University), and Yutaka Akiyama (Tokyo Institute of Technology) with three subthemes as shown in Figure 1. We have been collaborating with biomedical projects and scientists shown in the mid-left of Figure 1. Publicly available data used for our group are summarized in the mid-right of Figure 1. The contributions of 2014 fiscal year are summarized below.
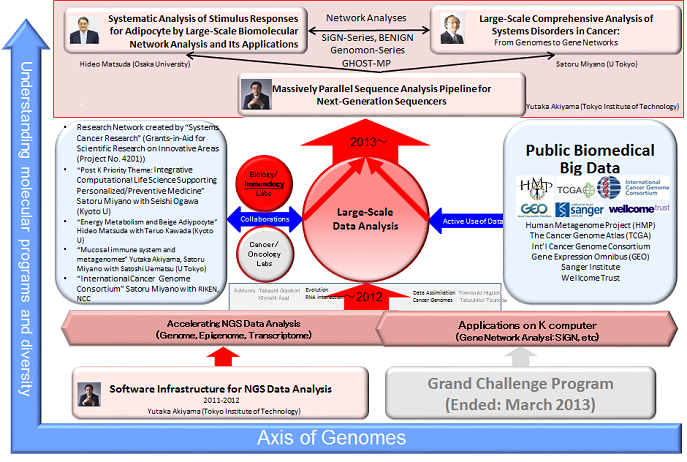
Figure 1: Group organization and subthemes.
1. Large-Scale Comprehensive Analysis of Systems Disorders in Cancer: From Genomes to Gene Networks (2013 – ) Satoru Miyano (The University of Tokyo)
Heterogeneity of cancer makes our understanding of cancer very difficult. Therefore, we need to analyze cancer data in large-scale. We analyzed the genomes and gene networks of several tens of thousands of cancer samples/cell lines. For the analyses, we used software applications “SiGN-Series” for gene network analysis, “Genomon-Series” for genomic sequence data and various bioinformatics tools, most of which have been developed in this project and the former project called the Grand Challenge Project for Life Science. We performed the world’s largest data analysis, including gene networks.
The first contribution is a high precision prediction of anti-cancer drug sensitivity/resistance and sensitivity-specific biomarkers by large-scale gene network analysis. The software application SiGN-L1 (developed in the Grand Challenge Program) estimates EACH gene network of EACH sample from gene expression profiles of many samples, by defining a modulator (e.g., IC50 score (drug resistance degree)). We used the Gene expression profile data of various cancer cell lines (728) and IC50 scores of 728 cancer cell lines for 142 drugs (IC50 score is the density of drug that reduces cancer cells by 50%) from “Sanger Genomics of Drug Sensitivity in Cancer”. We found that SiGN-L1 is not so robust agaist outliers and the Sanger data have many outliers. We improved the method by devising a robust kernel-based L1-type regularization. It is effective for outlier detection in high dimensional genomic data via PCA and effective for patient-specific analysis even in the presence of outliers. Gene network analysis was done again for 600 cancer cell lines and 101 drugs on K computer. Then our prediction method based on gene network data analysis exceeded the method by Garnett MJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012 Mar 28; 483 (7391) :570-575.
The second contribution is a discovery of a long non-coding RNA modulating an oncogene MYC. By analyzing 8,000 gene networks including non-coding RNAs (more than 10,000) from 8,000 lung cancer patient samples, our collaboration with Professor Takashi Takahashi (Nagoya U) led us to a discovery of novel long non-coding lncRNA MYMLER, named by Takahashi, that modulates MYC for selective target regulation. A 30-year mystery was solved with K computer power. More and more discoveries about non-coding RNAs are coming out now (not yet published). In this study, an improvement of SiGN-L1 (GIMLET) was developed to capture the relation among MYC, long non-coding RNAs, and target genes. Before K computer, we could not even imagine such large-scale network analysis.
The third is an on-going study based on a large-scale whole genome analysis of anaplastic thyroid cancer, a very rare cancer. This is a collaboration with Professor Koshi Mimori (Beppu Hospital, Kyushu University) and Shinya Uchino (Noguchi Hospital). Most of thyroid cancer is well-differentiated thyroid cancer, which shows the best prognosis among human cancers. However, it sometimes transforms anaplastic thyroid cancer, which shows the worst prognosis. Although accumulation of extra alterations in well-differentiated cancer presumably causes the anaplastic transformation, details of the causal mechanisms remain unclear. We performed whole gene sequencing of 12 anaplastic thyroid tumor and 6 paired-normal samples (provided by Noguchi Hospital, Beppu City, Oita Prefecture). We are identifying causal alterations for transformation from well-differentiated to anaplastic thyroid cancer (Figure 2), by jointly analyzing TCGA data of well-differentiated thyroid cancer.
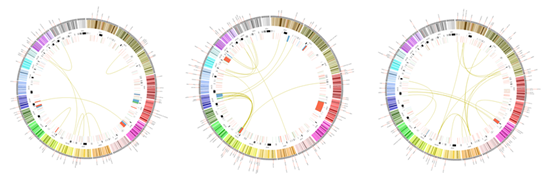
Figure 2: We found structural variations in anaplastic thyroid cancer.
2. Comprehensive Analysis of Gene Networks of Adipose Tissues Responding to Cold Exposure (2011 – ) Hideo Matsuda (Osaka University)We have been working on comprehensive analysis of gene networks of adipose tissue responding to cold exposure in collaboration with Professor Teruo Kawada (Kyoto University). Our study suggested new mechanisms of thermogenesis in beige adipose tissue. We developed a software application BENIGN for large-scale biomolecular network analysis, and applied it to comprehensive analysis of gene networks of adipose tissue responding to cold exposure. Beige adipocyte is transformed from white (normal) adipocyte upon cold stimulus (Figure 3). This process is called browning. The role of beige adipocyte is adaptive thermogenesis by dissipating energy stored in white adipocyte. Thus beige adipocyte is thought to be anti-metabolic cell against obesity. Our study revealed new mechanisms for transforming to beige adipocyte by large-scale biomolecular network analysis including microRNAs. We found a novel mechanism to control the transformation.
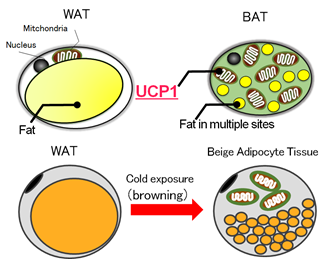
Figure 3: White adipocyte tissue (WAT), brown adipocyte tissue (BAT), and transdifferentiation from WAT to Beige Adipocyte Tissue
3. Massively Parallel Sequence Analysis Pipeline for Next-Generation Sequencers and Metagenome Analysis (2011 – ) Yutaka Akiyama (Tokyo Institute of Technology)
We have developed GHOST-MP, a parallel software application for large-scale metagenome analysis on K computer. The performance of GHOST-MP was tested using 80,000 nodes of K computer. A notable achievement in the development of GHOST-MP is a drastic reduction of I/O load.
For applications of GHOST-MP, we are collaborating with experts Professor Satoshi Uematsu (University of Tokyo and Chiba University) and Professor Kazuyuki Ishihara (Tokyo Dent Coll). Professor Uematsu is an expert on intestinal tract. We may reveal the interaction between host-genes and gut microbiota with knock-out mouse by metagenomic analysis using GHOST-MP. Virus metagenome analysis is also of our interest. Diarrhea by cholera or pathogenic E. coli is a major cause of babies and children’s death in developing countries. There exist people who have secretory IgA for cholera toxin B subunit (CTB), but have no history of cholera infection. We analyze microbiota of such people by GHOST-MP (Figure 4) and compare it with that of people who do not have secretory IgA for CTB; this has the potential to find microbiota that can be used for prevention to diarrhea. Our challenge is to find microbiota that can induce protective immunity to diarrhea caused by cholera or pathogenic E. coli. Professor Ishihara is an expert on oral bacteria. We may elucidate the differences between periodontitis patients and healthy individuals by metagenomic analysis using GHOST-MP
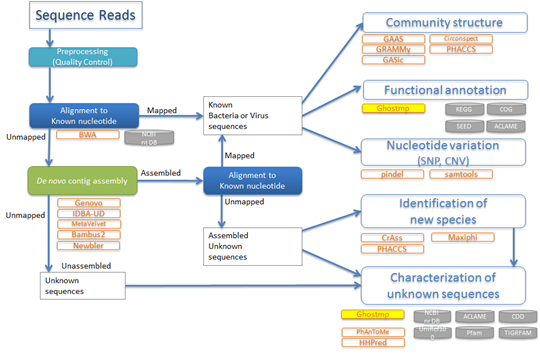
Figure 4: Metagenome Analysis Pipeline on K Computer.
- Large-Scale Comprehensive Analysis of Systems Disorders in Cancer: From Genomes to Gene Networks (2013 – ) Satoru Miyano (The University of Tokyo)
- Comprehensive Analysis of Gene Networks of Adipose Tissues Responding to Cold Exposure (2011 – ) Hideo Matsuda (Osaka University)
- Metagenome Analysis of Human Oral Microbiomes by World Deepest Homology Search (2011 – ) Yutaka Akiyama (Tokyo Institute of Technology)
Report on Research
Structural analysis of protein in solution by molecular dynamics simulation and small angle X-ray scattering
Yuichi Kokabu (Theme 1)
Computational Life Science Laboratory, Graduate School of Medical Life Science, Yokohama City University
Coarse-grained simulation : Investigation of the molecular mechanism of signaling pathway using CafeMol
Ryo Kanada (Theme 1)
Takada Laboratory, Department of Biophysics, Division of Biological Sciences, Graduate School of Science, Kyoto University
Exploring a chemical reaction in an intracellular crowded environment by the QM/MM method
Motoshi Kamiya (Theme 1)
Department of Chemistry, Graduate School of Science, Kyoto University
Development of a central nervous system model for elucidating the onset mechanism of Parkinson’s disease
Jun Igarashi (Theme 3)
Neural Computing Unit, Okinawa Institute of Science and Technology (OIST)
Multi-scale simulation of platelet aggregation in initial stage of thrombogenesis
Seiji Shiozaki (Theme 3)
Division of Cardiovascular Medicine, Department of Internal Medicine, Tokai University School of MedicinecTechnology (OIST)
Simulating cancer evolution to understand principles generating intratumor heterogeneity
Atsushi Niida (Theme 4)
Human Genome Center, Institute of Medical Science, The University of Tokyo





