Theme2 Simulation applicable to drug design
Simulation applicable to drug design

Group Leader:
Hideaki Fujitani
(Research Center for Advanced Science and Technology, The University of Tokyo)
Summary
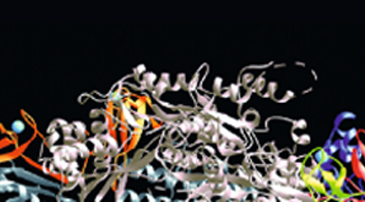
Since many drugs target proteins, we have to find a compound that interacts strongly with the intravital target protein (ligand) to discover a more effective drug. We aim at rapid and efficient development of low molecular weight drugs in the following manner. By use of molecular dynamics calculations using the K computer, we run a simulation of a system including a target protein and a drug candidate compound, investigate protein-compound interaction, and design a new compound which acts only on the target protein.
Approach during the Past Five Years and its Achievements
Publications
1. Hideaki Fujitani, Keiko Shinoda, Takefumi Yamashita, and Tatsuhiko Kodama:
“High performance computing for drug development on K computer“
J. Phys.: Conf. Ser. 454 012018 (2013)
2. Takefumi Yamashita, Akihiko Ueda, Takashi Mitsui, Atsushi omonaga, Shunji Matsumoto, Tatsuhiko Kodama, Hideaki Fujitani:
“Molecular Dynamics Simulation-Based Evaluation of the Binding Free Energies of Computationally Designed Drug Candidates: Importance of the Dynamical Effects”
Chemical and Pharmaceutical Bulletin vol.62 pp. 661-667 (2014)
3. Takefumi Yamashita:
“Improvement in Empirical Potential Functions for Increasing the Utility of Molecular Dynamics Simulations”
JPS Conf. Proc. 5, 010003 (2015)
4. Takefumi Yamashita, Akihiko Ueda, Takashi Mitsui, Atsushi Tomonaga, Shunji Matsumoto, Tatsuhiko Kodama, Hideaki Fujitani:
“The Feasibility of an Efficient Drug Design Method with High-Performance Computers“
Chemical and Pharmaceutical Bulletin,
vol. 63, pp. 147–155 (2015)
5. Nakayama, Mizohata, Yamashita et al.,:
“Structural features of interfacial tyrosine residue in ROBO1 fibronectin domain-antibody complex: Crystallographic, thermodynamic, and molecular dynamic analyses“
Protein Sciecne vol 24(3) pp 328-340 (2015)
Group organization

As of April 1, 2015