
Findings 2013
Pioneering the Future of Computational Life Science toward Understanding and Prediction of Complex Life Phenomena
Our Mission : BioSupercomputing Newsletter Vol.5 SPECIAL INTERVIEW
Development of R&D projects : BioSupercomputing Newsletter Vol.6 SPECIAL INTERVIEW
Program Director Toshio Yanagida
Deputy Program Director Akinori Kidera
Deputy Program Director Yukihiro Eguchi
Annual Report(Fiscal Year 2013)
- Theme 1 Simulations of biomolecules in cellular environments
- Theme 2 Simulation applicable to drug design
- Theme 3 Hierarchical integrated simulation for predictive medicine
- Theme 4 Large-scale analysis of life data
Simulations of biomolecules in cellular environments (Theme 1, Yuji Sugita, GL)
Currently, molecular dynamics (MD) simulations of biomolecules, like proteins, DNA, biological membranes, and so on, can be performed routinely on computers. One of the next big challenges in MD simulation community is to make dynamic models of a whole cell using multi-scale molecular models. Toward the whole cell modeling, we have attempted to perform large-scale simulations of proteins, DNA, and their complex under realistic cellular environments on K computer. The project contains the two subprojects: (i) the effect of macromolecular crowding on enzyme reactions in EGF signal pathway and (ii) multi-scale modeling of nucleosomes and chromatin. The following is the research summary of the two subprojects, which was done in the fiscal year 2013.
(1) The effect of macromolecular crowding on enzyme reactions in EGF signal pathway. To investigate the effect of macromolecular crowding on proteins, RNA, and metabolites, we simulated bacterial cytoplasmic systems based on the all-atom molecular models with explicit solvents. In this simulation, we used the GENESIS molecular dynamics package, which we have developed in RIKEN AICS. We also performed coarse-grained Brownian dynamics (BD) simulations of the same system with/without hydrodynamic effect. The protein diffusion in MD simulations is in good agreement with that in BD simulations with hydrodynamic interaction, although the simulation length of MD simulation was much shorter than that in BD simulations. We also examined protein conformational stability of proteins and RNA and found some proteins change their stability in cellular environments. Now, we are analyzing protein-protein, protein-RNA, protein-metabolites interactions based on the all-atom MD simulations. This information should be useful to make coarse-grained models on enzymatic EGF signal pathways (pSpatiocyte). In addition to the BD simulations, the group tested two other coarse-grained models: one is the single particle simulation and another is the coarse-grained Go-model simulation (CafeMol). The phosphorylation of MEK was also investigated using QM/MM hybrid free-energy calculations.
(2) Multi-scale modeling of nucleosomes and chromatin. In this subproject, we used multi-scale modeling approaches, which include coarse-grained MD (by CafeMol), all-atom/coarse-grained MD SAXS (MARBLE/CafeMol), and all-atom MD (SCUBA). By CafeMol, we could simulate a multi-nucleosome (for instance, a tri-nucleosome) to investigate how a chromatin is folded as a compact form in chromosomes. We also simulated functions of transcription factors in such nuclear crowding environments using CafeMol. To connect the simulation information with experimental data, we have started to develop all-atom/coarse-grained MD SAXS method. In this year, we applied this method to a single nucleosome in solution to compare the simulation structures with SAXS profiles. The protein-DNA interactions are investigated by performing large-scale free-energy calculations of a single nucleosome in explicit water. We compared the interactions between DNA and canonical histone with those between DNA and histone variants.
Simulation applicable to drug design(Theme 2, Hideaki Fujitani, GL)
The drug design is more costly and time-consuming in spite of the continuous progress of technologies, efficient and logical drug development methods are necessary. The computational structure-based drug design (SBDD) approach is one promising prescription. and therefore many methodologies and programs have been proposed. In any computational SBDD methods, drug candidates, which have high binding affinity with the target protein, are selected from the chemical compound libraries or the computationally designed compound groups. For the selection, it is necessary to evaluate the binding free energy for every compound. Thus, the accuracy of the binding free energy evaluation is critical to the success of the computational SBDD. If the compound group includes several high-affinity compounds but if the selection method does not work well, it is impossible to discover strong candidates. However, the standard computational SBDD methods introduce rough approximations into the binding free energy prediction in order to reduce the required computational resource; the target protein is assumed to be rigid and the water molecules are not taken into account explicitly.
We examined the approximate evaluation method in comparison with an exact molecular dynamics (MD) simulation-based method (named MP-CAFEE). It is important to note that MP-CAFEE can predict binding free energies accurately. In all MD simulations of the MP-CAFEE procedures, we employed the FUJI force field. The backbone torsion parameters of FUJI force field were determined to agree with LCCSD(T0)-level ab initio molecular orbital calculation results so that it can accurately describe the protein motion. We calculated the binding free energies for 58 selected drug candidates using MP-CAFEE with K computer. The compounds were generated by OPMF, a novel fragment-based de novo drug design method, and the ligand-protein interaction energy was examined as an example. The results showed that the correlation between the binding free energy and the interaction energy was not strong enough to clearly distinguish compounds with nM-affinity from those with μM-affinity. This implies that the accurate description of equilibrium states and the accurate evaluation involving entropic effects are necessary to predict the absolute binding free energy with the chemical accuracy.
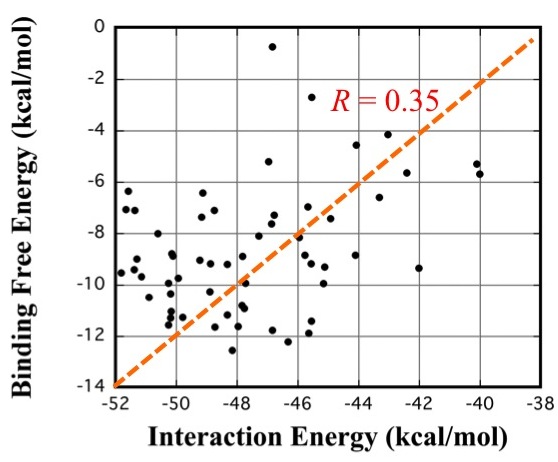
FIG. 1:Weak correlation between interaction energy and binding free energy
The simulation is corresponding to the brain size of small primate such as a marmoset. Using this neuron-network simulator NEST, the modeling of Parkinson’s disease brain was conducted. Doya’s group in OIST has been working on the modeling of basal ganglia, especially the influence coming from the reduction of dopamine supply. They showed that -band oscillation occurred in basal ganglia due to the lack of dopamine which was also observed in the monkey experiment of Parkinson’s disease.
This basal ganglia model for Parkinson’s disease needs to be connected with the other parts. The unified model of “Basal Ganglia” – “Thalamus” – “Cortex” – “Spinal Cord” – “Musculoskeletal System” has been under the development.
Hierarchical integrated simulation for predictive medicine (Theme 3, Shu Takagi, GL)
We have been working on mainly 3 types of software development. One is for Brain-Nervous-Musculoskeletal coupling method to predict the pathological condition of Parkinson’s disease. The other two is related to cardiovascular systems. One is for multiscale thrombosis simulator and the other is a heart simulator which has been already well-developed and known as UT-Heart.
In the fiscal year of 2013, as a starting point of large scale brain simulation, the largest neuronal network simulation was achieved using the software NEST on K computer. Dr. Igarashi in Doya team (OIST) is a main contributor of Japanese side, collaborating with Dr. Mark Diesmann and Abigail Morrison in the Institute of Neuroscience and Medicine at Jülich. They succeeded in simulating a network consisting of 1.73 billion nerve cells connected by 10.4 trillion synapses using the entire system of K-computer, which has 82,944 processors. The scalability of the simulation is shown in Fig.1 and also in the following website.
http://www.riken.jp/en/pr/press/2013/20130802_1/
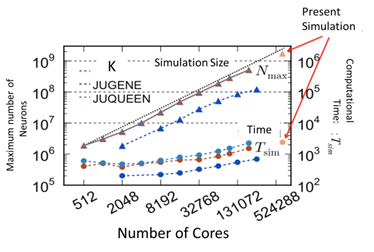
Fig. 1 Large Scale Neuronal Network Simulation using K-computer
The simulation is corresponding to the brain size of small primate such as a marmoset. Using this neuron-network simulator NEST, the modeling of Parkinson’s disease brain was conducted. Doya’s group in OIST has been working on the modeling of basal ganglia, especially the influence coming from the reduction of dopamine supply. They showed that β-band oscillation occurred in basal ganglia due to the lack of dopamine which was also observed in the monkey experiment of Parkinson’s disease. This basal ganglia model for Parkinson’s disease needs to be connected with the other parts. The unified model of “Basal Ganglia” – “Thalamus” – “Cortex” – “Spinal Cord” – “Musculoskeletal System” has been under the development.
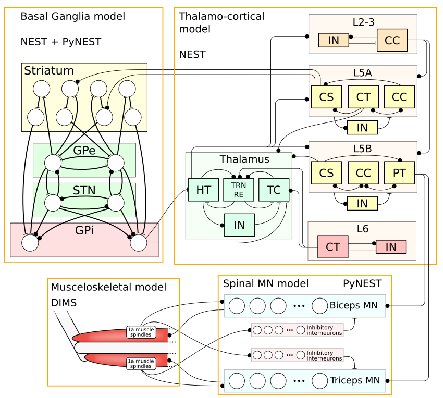
Fig.2 Unified model of “Basal Ganglia” – “Thalamus” – “Cortex” – “Spinal Cord” -“Musculoskeletal System”
Development of skeletal muscle model from muscle fiber level and its coupling to neural system was conducted under the collaboration of Takagi and Nakamura’s team. Takagi’s team developed detail multiscale modeling of skeletal muscle from muscle fiber modeling and Nakamura’s team worked on the modeling of entire human body from the coarse-grained muscle fibers (Fig.3).
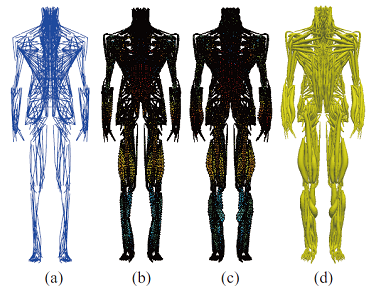
Fig. 3 Automatic reconstruction of volumetric musculoskeletal systems developed in Nakamura’s group
For the multiscale-multiphysics heart simulator, UT-Heart, 3-stage hierarchical integration as achieved from Sarcomere dynamics, through muscle cells, to the whole heart. It is the world’s first achievement in the sense that sarcomere dynamics is simultaneously coupled to the entire heart motion. The transition state theory with Monte Carlo method was employed to simulate the cross-bridge behavior of actin-myosin protein interactions. The present transition state models are shown in Fig. 2. Through this 3-stage integrated model, it was shown that the cooperative behavior of the myosin molecules is crucial for the normal behavior of the entire heart. The snapshot is shown in Fig.4 and the detail movie is available at the following website of our project.
http://www.scls.riken.jp/research/03_integration/
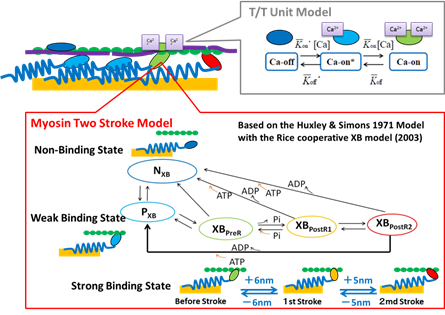
Fig. 4 Molecular Scale Stochastic Model for Sarcomere Dynamics
For the multiscale thrombosis simulator, we have continued developing the model to simulate the efficacy of antiplatelet drags. As antiplatelet drugs, we have been making the two types of models. One is the P2Y12 inhibitor which is represented by Clopidogrel, and the other is GPIIb/IIIa inhibitor. Corresponding experiments were also conducted by the research group of Goto (Tokai Univ.). Fig. 5 illustrates the large scale simulations, which is the same size of the flow chamber experiments. The simulations were conducted for the platelets adhesion process and qualitatively good agreement between experiment and the simulation was achieved that is, platelets do not adhere on the wWF immersed chamber wall without presence of RBCs.
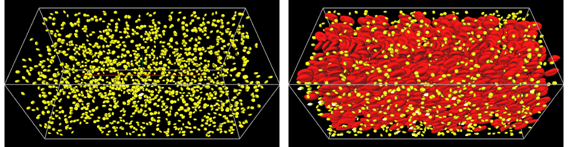
Fig.5 Large scale simulation for the platelets adhesion process on the vWF immersed wall hematocrit. Effect of Hematocrit Value (Ht)
Large-scale analysis of life data (Theme 4, Satoru Miyano, GL)
In order to focus on more K computer-oriented subthemes which should mutually enhance and create synergy, the group was reorganized. Satoru Miyano remained as the group leader and two PI’s Yutaka Akiyama (Tokyo Institute of Technology) and Hideo Matsuda (Osaka University) continued their subthemes. Until March of 2013, Miyano’s mission was to manage this group, but from April of 2013, Satoru Miyano joined and started the research. Our biomedical targets are cancer, obesity, and human metagenome. Recent studies revealed that all these three targets are mutually related in our diseases (Yoshimoto S et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 499(7456):97-101, 2013). The group is organized as shown in Figure 1. We summarize the development and contributions of 2013 fiscal year.
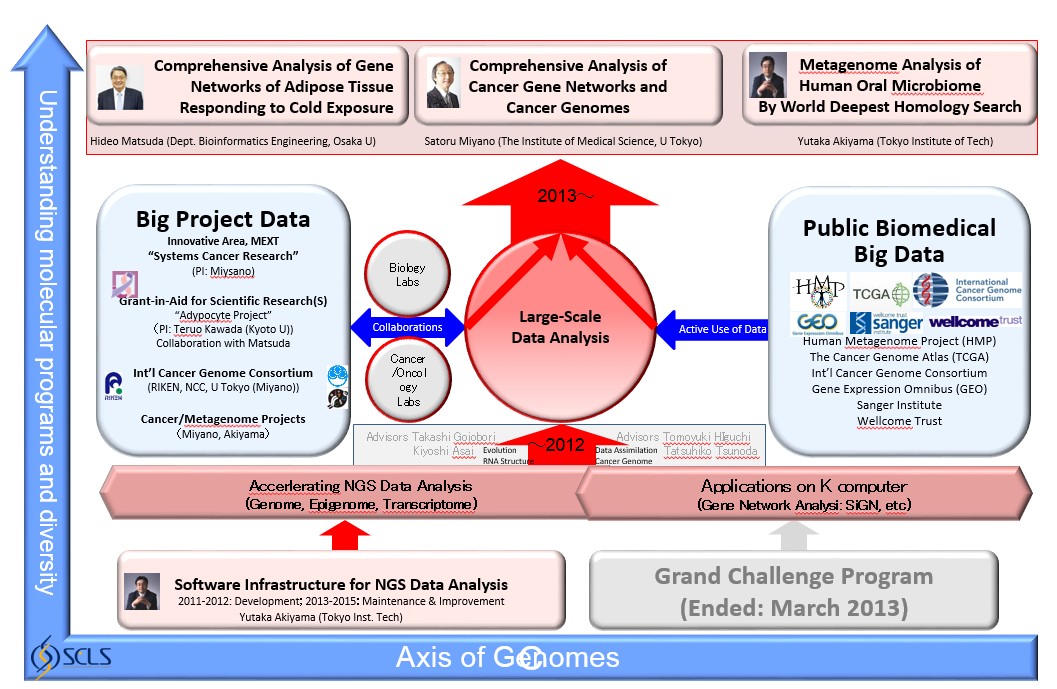
Figure 1: Group organization and subthemes.
- Large-Scale Comprehensive Analysis of Systems Disorders in Cancer: From Genomes to Gene Networks (2013 – ) Satoru Miyano (The University of Tokyo)
- Comprehensive Analysis of Gene Networks of Adipose Tissues Responding to Cold Exposure (2011 – ) Hideo Matsuda (Osaka University)
- Metagenome Analysis of Human Oral Microbiomes by World Deepest Homology Search (2011 – ) Yutaka Akiyama (Tokyo Institute of Technology)
Report on Research
Free Energy Profile Calculations for Changes in Nucleosome Positioning with All-Atom Model Simulations
Quantum Beam Science Directorate, Japan Atomic Energy Agency
Hidetoshi Kono, Hisashi Ishida, Yoshiteru Yonetani, Kimiyoshi Ikebe
(Theme 1, Yuji Sugita, GL)
Simulation Applicable to Drug Design
Research Center for Advanced Science and Technology, The University of Tokyo
Hideaki Fujitani
(Theme 2, Hideaki Fujitani, GL)
Estimation of Skeletal Muscle Activity and Neural Model of Spinal Cord Reflex
Information Science and Technology, The University of Tokyo
Yoshihiko Nakamura
(Theme 3, Shu Takagi, GL)
An Ultra-fast Analysis System for Next-Generation DNA Sequencer Data
Graduate School of Information Science and Engineering, Tokyo Institute of Technology
Yutaka Akiyama, Takashi Ishida, Masanori Kakuta, Shuji Suzuki
(Theme 4, Satoru Miyano, GL)





