
Findings 2011
Annual Report(Fiscal Year 2011)
- Theme 1 Simulations of biomolecules in cellular environments
- Theme 2 Simulation applicable to drug design
- Theme 3 Hierarchical integrated simulation for predictive medicine
- Theme 4 Large-scale analysis of life data
Simulations of biomolecules in cellular environments (Theme 1, Yuji Sugita, GL)
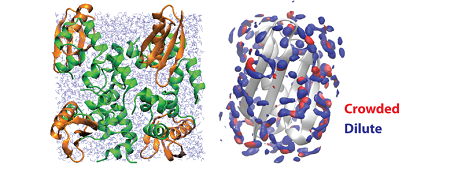
To simulate biomolecular dynamics and functions under cellular environments, we need novel multi-scale/multi-resolution software. In this project, we are developing three programs, GENESIS, SCUBA, and pSpatiocyte. Both GENESIS and SCUBA allow us to simulate all-atom molecular dynamics (MD) simulations for studying conformational dynamics of proteins and nucleic acids in cellular environments. In contract, pSpatiocyte can perform large-scale cellular simulations based on the coarse-grained diffusion-reaction model. Currently, these programs are shown to scale up to several thousand CPUs in K computer. We are trying further to improve the performance in K computer. We also develop analysis program (Motion-Tree) for large-conformational changes in proteins or nucleic acids, which are expected in long MD simulations. Due to the limited CPU time in K computer, we could not perform long production simulations. We therefore performed biomolecular simulations in cellular environments using conventional supercomputers in RIKEN, JAEA, and the University of Tokyo. One of the highlights in the last financial year (2011) was hydration of proteins in crowded environments. In this study, the effect of protein crowding on the structure and dynamics of water was examined from explicit solvent MD simulations of a series of protein G and protein G/villin systems at different protein concentrations. We analyzed the hydration structure in terms of radial distribution functions, three-dimensional hydration sites, and preservation of tetrahedral coordination. Analysis of hydration dynamics focused on self-diffusion rates and dielectric constants as a function of crowding. We observed significant changes in both structure and dynamics of water under highly crowded conditions. The structure of water is altered mostly beyond the first solvation shell. Diffusion rates and dielectric constants are significantly reduced following linear trends as a function of crowding reflecting highly constrained water in crowded environments. The results from this study suggest a prescription for modeling solvation in simulations of cellular environments.
Publications
1. Ryuhei Harada, Yuji Sugita, and Michael Feig:
”Protein Crowding Affects Hydration Structure and Dynamics“
J. Am. Chem. Soc. (2012) 134, 4842-4849.
2. Michael Feig and Yuji Sugita:
”Variable Interactions between Protein Crowders and Biomolecular Solutes are Important in Understanding Cellular Crowding“
J.Phys. Chem. B, (2012) 116 (1), 599–605.
Simulation applicable to drug design (Theme 2, Hideaki Fujitani, GL)
We usually perform 384 molecular dynamics simulations with different parameters in MP-CAFEE to obtain a free energy. If we use 10 CPU cores for each molecular dynamics simulation, we need 3,840 CPU cores in total. We do not have such a big computer in our laboratory but K computer has more than 88 thousands of nodes (704 thousands of CPU cores). We can perform a MP-CAFEE calculation as a single job on K computer. We implemented such function to our program. Using a typical pharmaceutical target protein in water we observed 95 % strong scaling between 3,840 nodes (30,720 CPU cores) and 1,920 nodes in the MP-CAFEE calculation on K computer. It is good parallel efficiency.
We can identify drug candidates with high binding affinity to the target protein by MP-CAFEE. When we get one binding affinity by two hour MP-CAFEE calculation with 3,840 nodes, we can calculate 275 molecule’s affinities in one day with the whole K computer. Although this number is attractive from drug development point of view, it is far from covering the diverse chemical space of the organic compounds.
We use structure based drug design methods to select and design the molecules for the MP-CAFEE calculations on K computer. One is de novo design method: OPMF (Optimum Packing of Molecular Fragments), which is proprietary software of FUJITSU. Others are commercial software for the structure based drug design from OpenEye, Accelrys, and Schrödinger. Using these tools we are designing new drug candidate molecules for the pharmaceutical target protein like PR-SET7. Then we perform molecular dynamics equilibration in water and MP-CAFEE calculations.
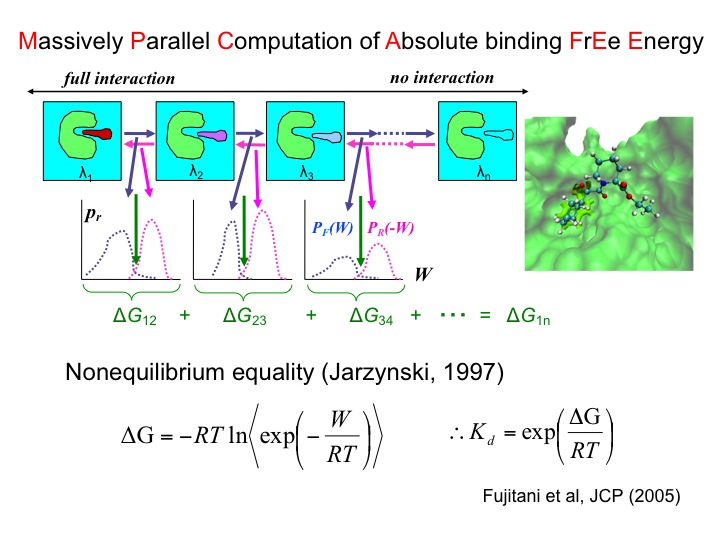
- MP-CAFEE (Massively parallel computation for absolute binding free energy)
- An epigenome therapy target protein: PR-Set7
Hierarchical integrated simulation for predictive medicine (Theme 3, Shu Takagi, GL)
1) Modeling of the activation of platelets and the efficacy of anti-platelet drug toward the numerical simulations of myocardial infarction.
Using the multiscale thrombosis simulator, which we have been working on (see Fig.1), we have started to build up the numerical model for the complicated chemical reaction triggered after the initial activation of the platelets, i.e., after the adhesion of the platelet on the injured vessel walls through the molecular bindings between GP1ba and vWF proteins. More specifically, we have worked on the modeling of anti-platelet drug: Clopidogrel. Fig.2 illustrates the bio-chemical reactions related to the drug efficacy of Clopidogrel (CLO) and the numerical results using this model are shown in Fig.3. The results indicate that CLO inhibits the activation process and the activated rate of P2Y12 and GPIIb/IIIa are reduced. This model will be extended to include more complicated biochemical process and will be coupled with the blood flow solver in the year of 2012.
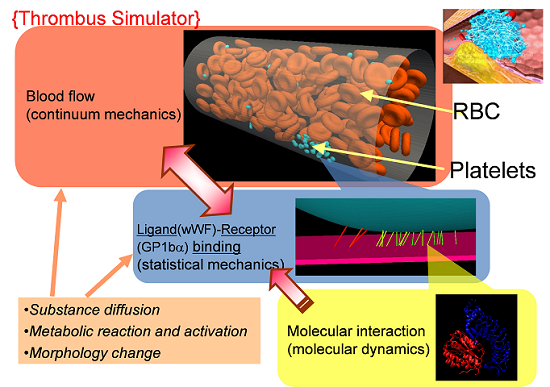
Fig.1 Schematic figure for the concept of multiscale thromobosi simulator
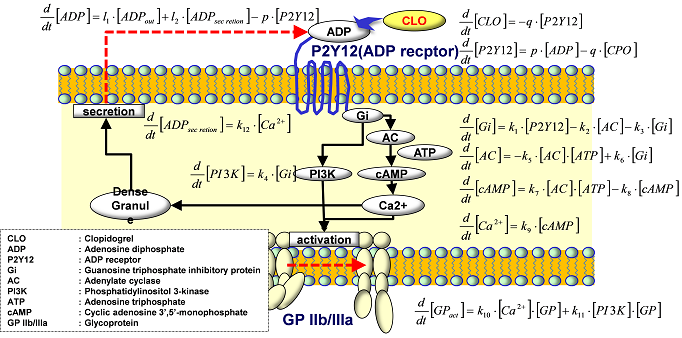
Fig.2 Present model of the efficacy of anti-platelet drug: Clopidogrel.
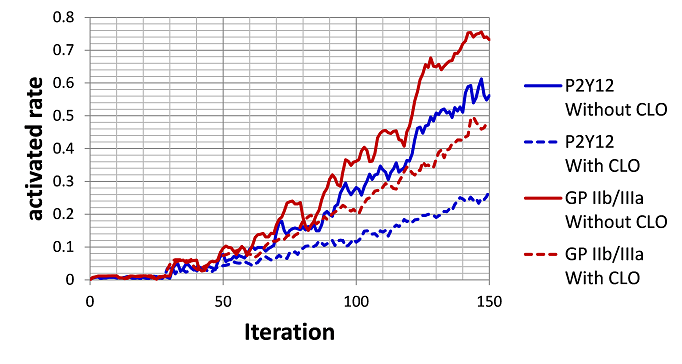
Fig.3 Simulation Results of efficacy of Clopidogrel (CLO)
2) Hierarchical modeling of motor dysfunction due to the neurological disorder: Modeling of Parkinson Disease,
This year, we have been working on the modeling of the Parkinson Disease (PD) based on the previous studies by Cutsuridis(2011) and Cesi (2008). Our model has a hierarchical structure, which couple the brain signals produced through the simulation of brain neural network system with the skeletal muscle contraction caused by the contraction of each muscle fiber through the biomechanical simulation. That is, by passing the spike signals from the brain to neuron pool at spinal code with the suitable feedback model, and relating them to the signals of motor neuron, we can have the muscle contraction model for the PD, since it is thought that abnormal brain signals is the cause of the abnormal muscle contraction such as the rigidity or tremor. We have started modeling of this complicated hierarchical system.
For the brain system, Doya’s research group in OIST has started using the brain neural network simulator (NEST) to model the abnormal brain signal caused by PD. The model is under the development and the simulation is going to be conducted in “K” computer in the year 2012. For biomechanical simulation of skeletal muscles, Takagi’s group in the University of Tokyo has been developing the method to conduct the muscle contraction process caused by the signal from the motor neuron. The results of continuum mechanical simulations of muscle contraction in lower leg part are shown in Fig.4. This simulation will be coupled with the generated brain signals. Nakamura’s group in the University of Tokyo has been also working on the biomechanical modeling and developing the method to include the muscle volume effect on the whole-body neuromuscular-skeletal system.
References
1) Vassili Cutsuridis, Origins of a repetitive and co-contractive biphasic pattern of muscle activation in Parkinson’s disease, Neural Networks, 24 (2011), pp.592-601.
2) Rogerio R. L. Cisi & André F. Kohn, Simulation system of spinal cord motor nuclei and associated nerves and muscles, in a Web-based architecture, J. Comput. Neurosci, 25(2008), pp.520-542.
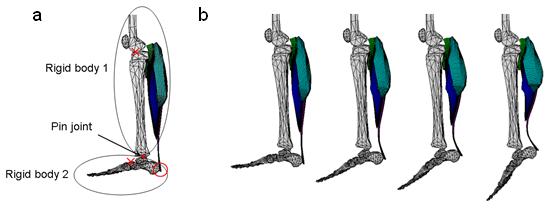
Fig. 4 Simulation results of the ankle joint movement generated by the activation of the triceps surae muscle. (a) Ankle joint model consist of rigid bodies and pin joint. (b) Ankle joint movement during the activation of the triceps surae muscle.
Large-scale analysis of life data (Theme 4, Satoru Miyano, GL)
- General management
- Development of data processing system for next-generation sequencer data analysis
- Development of methods for predicting RNA-RNA interaction and comprehensive analysis of RNA data
- Development of large-scale biomolecular network analysis techniques
- Metagenomic analysis and comparative genome analysis research





