分子動力学計算とX線小角散乱実験による
タンパク質の溶液構造解析

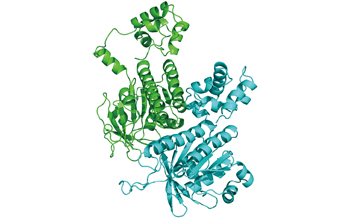
図1:MDシミュレーションによって予測されたRad51二量体の構造
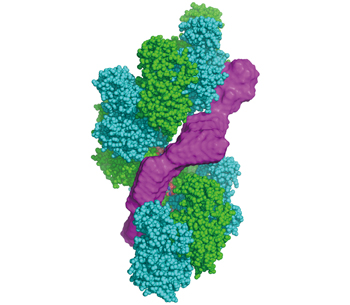
図2:Rad51フィラメント(緑とシアン)の溝にSwi5-Sfr1の溶液構造(マゼンタ)が結合したモデル
タンパク質などの生体高分子は溶液中に存在します。溶液中のタンパク質は、1つの決まった構造を取っているのではなく、その構造は熱運動によって揺らいでいます。このため、溶液中のタンパク質の揺らぎを含んだ構造を決定することは生体内でのタンパク質の機能を理解する上で重要です。今回、タンパク質の溶液構造を決定(もしくは予測)する方法として分子動力学(MD)シミュレーションとX線小角散乱法(SAXS)を、最近の我々の研究成果を例にとって紹介します。
MDシミュレーションは、溶液中のタンパク質の挙動について、運動方程式を解くことによって再現する手法です。この手法により、全原子レベルでタンパク質の挙動を予測できます。我々は相同組換えにおける鎖交換反応を触媒するRad51タンパク質フィラメントに関して、その最小単位である二量体のMDシミュレーションを行い、Rad51フィラメントの溶液中の構造を予測しました(図1)。
一方、SAXSは溶液中のタンパク質に直接X線を照射し、得られた散乱パターンから分子の概形を測定する手法です。解像度は低いけれど、熱揺らぎを含んだ溶液中の構造を測定できます。今回、我々はSAXSを用いて、Rad51の活性化因子であるSwi5-Sfr1複合体(S/S)というタンパク質の溶液構造解析を行いました。その結果、S/Sは溶液中で非常に細長い構造を取っている事が分かりました。
このS/Sの細長い構造は予測したRad51フィラメントの溝の部分に非常に良く適合しました(図2)。このことからRad51フィラメントはその溝部分にS/Sが結合することによって活性化されるという仮説を立て、その検証を進めています。
MDシミュレーションは原子レベルの解像度での揺らぎを含んだ構造を予測できますが、実験的な検証が存在すれば確信度が高まります。そこで我々は、MDシミュレーションの結果が妥当かどうかを、SAXSの実験データと比較することにより判断できないかと考え、MDシミュレーションの結果から、理論的にSAXSの散乱パターンを計算する手法を開発しました。MDシミュレーションから計算したSAXSパターンとSAXSの実験データが一致すれば、そのMDシミュレーションは妥当であると判断できます。実際にこの手法をSfr1のN末端から177残基が欠損した変異体Swi5-Sfr1dN177に適用し、原子レベルでの揺らぎを含んだ溶液構造を解析しました。
現在、ヌクレオソームというDNA結合タンパク質に対して、この手法を適用しています。ヌクレオソームは巨大な分子であり、全原子MDシミュレーションでは計算時間が掛かりすぎるため、1つのアミノ酸を1つの粒子に置き換えて計算する粗視化MDシミュレーションを用いて、ヌクレオソームのDNA末端の揺らぎとSAXSの散乱パターンの解析を進めています。