最先端のシークエンサーをはじめさまざまな先進的計測技術が、生命システムデータの精緻化・大規模化を加速している。課題4「大規模生命データ解析」(代表:東京大学医科学研究所・宮野悟教授)は、「京」に最適化した最先端・大規模データ解析の基盤を構築し、生命プログラムの複雑性や多様性、さらには進化を理解するとともに、ゲノムを基軸とした生体分子ネットワーク解析研究を推進している。最新の情報処理技術を活用したバイオインフォマティクス(生命情報学)研究の最前線で奮闘する3名の研究者に話を聞いた。
バイオインフォマティクスとの出会い

図:(左上)シミュレーションによって得られた腫瘍
図:(右上)ゲノム変異のプロファイル
図:(下)時系列で可視化した腫瘍の進化
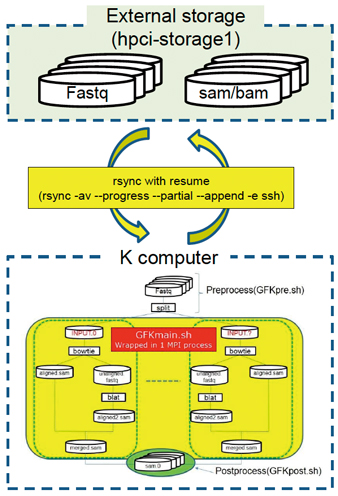
図:がんのシステム異常をシークエンス、トランスクリプトームレベルで大規模網羅的に明らかにするソフトウェアGenomon-Fusion for K computerの概念図。個別にMPI化すると同期処理で時間が無駄になるため、全体を1つのMPIプロセス化するなど、「京」への移植にはさまざまな改良が加えられた。
●みなさんは、どのような経緯でバイオインフォマティクス分野の研究に携わるようになったのですか。
新井田 学部では、がんについての分子生物学的な実験をやっていました。ちょうど大学院に入るころにヒトゲノムプロジェクトが終わり、ゲノム科学に触れて、バイオインフォマティクスに興味を持つようになりました。もともとコンピュータはあまり得意ではありませんでしたが、自分なりに勉強を続け、大学院を出てから宮野先生の研究室に来て、バイオインフォマティクスの方法論などに取り組むようになりました。そして、宮野先生から「『京』のプロジェクトがあるからやってみないか」と誘われたのです。自分では、まさか日本一のスーパーコンピュータを使うようになるとは思ってもいませんでしたが、伊東さんたちの助けを借りつつ研究を進めています。
伊東 私は工学部の出身で、専門は流体。HPCI戦略プログラムでいえば分野4のものづくり分野に関連する研究をやっていました。ものづくりの分野ではコンピュータの利用が盛んで、私もスーパーコンピュータを使うシミュレーション研究をずっとやっていたのですが、すでに産業分野でどんどん利用されており、細分化というか先鋭化して、個別の案件に近い研究が進められています。自分としては、もっと違うことがやってみたいという気持ちでした。そんなときに宮野先生からお誘いいただき、昨年4月にこちらの研究室に来ました。まだバイオ分野はよく分かっていないところがあり、新井田さんをはじめみなさんに指導を受けています。逆に「京」での並列化や最適化の部分では、研究室の方々はまだ慣れていない面もある ので、その部分を私がサポートする感じで、協力しながらやっています。
角田 学部時代は理学部でしたが、卒業した後に農学部の応用工学科に進み、初めてコンピュータを使った研究をするようになりました。そこではタンパク質間相互作用について、あとはタンパク質の立体構造の解析や予測などの研究をしていました。大学院を修了した後も1年半ほど研究室に残りましたが、2011年の半ばころに秋山研究室に移り、課題4のプロジェクトに参加することになりました。HPCそのものにも興味がありましたが、それよりも、規模の大きな計算に取り組むことで、研究の幅が広がるのではないかという思いでした。
がんのシミュレーション研究にも取り組む
●課題4のプロジェクトで、みなさんは実際にどのような研究に取り組んでいるのですか。
新井田 宮野研究室はがんの実験のラボや医師たちといろいろな連携研究をしています。そこから大量のデータが得られるのですが、それを迅速に解析することが急務になっています。今やっている研究の1つは、遺伝子セット情報に基づいてmRNA発現データ中で共発現している遺伝子群、発現モジュールを抽出するEEM(Extraction of Expression Module)というソフトウェアを使って連携研究先のデータを解析し、実験と合わせてがんの転写プログラムを明らかにするというものです。EEMの「京」への移植については、伊東さんらに助けてもらっています。もう1つも連携研究から出てきたがんのシミュレーションの研究です。がんのDNAはそれぞれの患者さんごとに違っているということは良く知られていますが、近年一人の患者さんの1つの腫瘍内でも、異なる場所のDNAを調べると、かなり多様であることが分かってきました。がんは、最初1つの正常細胞が変異を蓄積し、さらに違う変異を持ったクローンが分岐し、最終的に腫瘍を見たとき、そこには全く違う遺伝子の変異を持つサブクローンがたくさん存在することが明らかになっています。実際、連携研究で大腸がんのマルチサンプリング、つまり1つの腫瘍内の違う場所からDNAを採ってシークエンスすると、まったく違う遺伝子のプロファイルが出ます。それはどうしてなのか、この疑問を解き明かすためにシミュレーションをして、進化のメカニズムを探ってみたいと考えています。シミュレーションのモデルは、伊東さんに「京」で動くようにしてもらい、いろいろなパラメーターで走らせることで、どのパラメーターが腫瘍内の不均一性をつくるのに重要かということを探索しているところです。将来的には、どういう薬が効くのかも含めてシミュレーションでがんの一生をたどり、治療戦略に活かしたいと考えています。
●「京」へのソフトウェアの移植ということで、研究には伊東さんも関わっているのですね。
伊東 そうですね。新井田さんだけに限らず、宮野研究室のなかで「京」を使える研究者はまだ少ないので、これまで使っていたソフトウェアを「京」で運用するためのサポートが、いちばん最初の私の仕事です。例えば、あるソフトウェアを「京」で大規模に動かしたいという希望が新井田さんから上がってきたら、その内容を私の方で解析して、「京」にそのまま移植できるか判断し、無理ならば何か別なものに代替するか、あるいは直接プログラムを書き換えて移植が可能な状態にします。さらに並列化もしないといけないので、そこまで作業を行って、新井田さんが使える状態に持っていくということです。
新井田 やはり「京」は、自分にとってはややハードルが高い部分があります。「京」は天文や物理など、昔からHPCを使っていた人向けにつくられていて、バイオの研究者には使いにくい部分があるように思えます。
伊東 確かに難しい部分はあります。バイオインフォマティクス分野もいろいろなソフトウェアが出ていますが、それらは1個のプログラミング言語で書かれているのではなく、パイプラインという形で、複数のソフトウェアを組み合わせているものが多いのです。例えばC言語で書かれたものの間にRの統計言語が入っていたり、何かスクリプトで処理が入っているなど、いろいろなものが入っていて、個別に分解しないとなかなか移植できないことも多いのです。また、バイオインフォマティクス分野でスパコンを使っているユーザーはそれなりにいますが、並列化の経験がある人は少ないと思います。そのため、いきなり「京」というのはハードルが高いかもしれません。
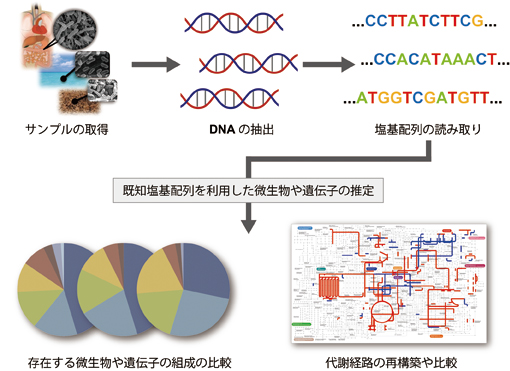
図:系統解析や機能解析を活用したメタゲノム解析の模式図
超高速で大規模な解析を可能にしたい
角田 私は、主に「京」を使ったメタゲノム解析パイプラインの開発を行っていて、パイプラインを使った実データの解析なども行っています。メタゲノム解析は、ある環境中に存在する微生物集団のゲノムを解析する手法です。環境中にはさまざまな種類の微生物がいますが、それらを混ざった状態のまま解析することで、どのような微生物が、どのような場所に、どのくらいいるのかを解析することができます。さらに、存在する割合などを見ることによって、環境と微生物がどのように相互作用しているかも調べることができます。ただ、1つの微生物ゲノムのシークエンシングと比べ、メタゲノム解析で扱う配列データのサイズは非常に大きくなります。その大量のデータを解析するために、「京」を使って大規模な計算を行うことで解決しようというのが、私のかかわっているプロジェクトです。メタゲノム解析パイプラインは、一連の処理を通して次世代シークエンサーから得られる配列断片と、既知配列データベースとの比較を行うことで、サンプルデータ中に含まれる遺伝子や機能の相対存在度に基づいた解析が可能です。そして、解析パイプラインで中心的な役割を担っているのが、GHOST-MPという並列相同性解析ソフトウェアです。1つのプロセッサの上で実行されるプログラムを大学院生がつくり、私はその逐次版のプログラムを「京」の上で並列実行させるプログラムに変え、大規模な解析を可能にするところを担当しています。私たちは、より解析精度を上げた計算をめざしていますが、そのためにはどうしても計算時間がかかってしまいます。それを減らすために、逐次版の高速化や高い並列度で効率よく動作させる手法に取り組み、両面から超高速かつ大規模な解析を可能にしようと試みています。
※以下太字で表記している箇所はWeb版でのみ公開のロングバージョン部分です。
●「京」を大規模データ解析に活用することの難しさも実感
━研究を進めていく上での難しさ、もしくは現在の課題は何ですか。
新井田 HPCIのプロジェクトに関わっていながら、こんなことをいうのはいかがかとも思いますが、「京」の扱い方については、まだ伊東さんに教えていただきながら進めている状況でして、もっと勉強しなければと思っています。並列化といったテクニック的なこともそうですが、スーパーコンピュータにもそれぞれ癖というか個性のようなものがあって、早く「京」に慣れなければということを実感しています。例えば、シークエンスデータを解析する場合、とにかくデータ量が膨大で、医科学研究所のスーパーコンピュータは大量のデータを扱うことに特化されている部分があるのですが、これを「京」で扱うためにいろいろと苦労しています。次世代スパコンでは、ぜひバイオ分野というか、大量データにも対応できるものにしてもらいたいと思います。
伊東 その点は、私も実感しています。確かに「京」では、計算するためのデータを準備するところがいちばんたいへんです。ちょうど今、私が最も時間を使って取り組んでいるのが、CCLE(米国Broad Institute)などのデータベースで公開されている大規模なヒト検体データを処理するシステムを構築するための、Genomon-Fusionというデータ解析パイプラインの「京」への移植です。計算のCPU時間という点では、「京」のフルノード換算で1日かからない程度なのに、実際に解析データを準備してから終了するまでに3カ月ほどかかっています。つまり、計算するより前のところに時間がかかっていて、これをどう解決したらよいのか、いろいろと悩んでいるところです。
新井田 最初にヒトゲノムプロジェクトの話をしましたが、このときは十数年かけて1人分を読んだわけです。ところが今ではシークエンサーの発達によって、数日で結果が出る時代です。データがどんどん出てきて、それを解析するためにはどうしても「京」のような大きなマシンが必要です。しかし、あえて申し上げると、「京」は大量データにスムーズに対応できるスペックではないので、そこをどうするかが課題かなと思います。
伊東 例えば、ソフトウェアが入っていないということなら、自分たちで補うことができますが、システム的にどうしても入らないものもあるので、そこはシステム側で対応してもらう必要があります。あと、先ほど申し上げたデータの問題は、ストレージの容量という観点からも厳しいものがありますし、さらにデータ転送ということについても厳しい部分があります。特に公開されているデータベースからデータをダウンロードするときに、特殊なソフトウェアを使わないと落とせないものが多いのです。そのために、一度こちらのスーパーコンピュータにダウンロードしてから「京」に移して解析を行い、それをもう一度こちらに戻すという手間がかかっています。実はこのあたりは当初全く想像していなくて、移植さえ終えれば後はどんどん解析できると思っていたのですが、実際はデータのハンドリングに大きな課題があって、これをどう解決したらよいのか考えています。研究に活用してもらうためには、研究者にとっての使い勝手のよさも重要です。専門知識を持った技術者が使えるレベルよりもさらにブラッシュアップして、新井田さんたち研究者が簡単に使えるようになっていなければいけないわけです。そこを考えると、まだまだ課題が多いと思っています。
角田 私も伊東さんと同じで、やはりデータの転送がいちばん面倒で、どうしても時間がかかってしまいます。そこが速くできないので、いくら並列度を高めても解消できない部分が残ってしまう。どうしたらいいのかと悩んでいるところです。
新井田 課題という話題からは外れますが、もともと自分は生命科学の分野にいて、伊東さんのようなHPCの専門家たちとは全く違った世界にいたのに、このプロジェクトに参加して、今一緒に研究をやっているということが非常に面白いと実感しています。このプロジェクトがなかったら、伊東さんや角田さんと出会えなかったでしょうし、違う専門分野を持った人たちが同じテーブルについて、お互いに知恵を出し合っていくことの楽しさや、そこで受けるカルチャーショックみたいなものを感じています。その意味でも、すごく面白いプロジェクトだと思います。
角田 私もスーパーコンピュータを扱い始めたのは、このプロジェクトに関わってからで、それまでは小規模なクラスターを扱う程度でした。最初はどう扱っていけばよいのか分からないことも多く苦労もしましたが、今は生命科学と計算科学の橋渡しができるようになりたいと思っています。今後は、この両者が結び付くことが不可欠になるはずですから。そのためには、自分自身がどちらも中途半端にならないように気をつけないといけませんね。
バイオインフォマティクスを医療に役立てたい
●今後の研究を通して、達成したいことは何ですか。
新井田 今やっているシミュレーションの仕事では、単にパラメーター探索などにとどまらず、実験データと照らし合わせて、そこからがんの進化原理であったり、多様性が出るメカニズムなどを見つけ出し、がん研究の新しいコンセプトを打ち出せる、そういうゴールをめざしていきたいと思っています。そのためには、もちろんシミュレーションの改良も大切ですが、やはり現場のお医者さんとコミュニケーションを取り、そこから新しい知識を得たり、実験データをうまく自分の解析にフィードバックするといったことが重要と考えています。そして将来は、バイオインフォマティクスを使って、医療に役立つ発見をしたいですね。
伊東 まずは、すでに公開/制限付き公開されているデータベースから、バイオロジストが生物学的な知見を得られる状態に処理するパイプライン開発とそれを用いた計算の実施です。それから、次世代シークエンサーのテクノロジー進歩が非常に速く、数年後には全く違うアプローチが必要になるかもしれません。そのような状況にも対応できるよう、スーパーコンピュータの特性とバイオインフォマティクスの要求をうまく融合させたソフトウェアの開発を進めていきたいと考えています。
角田 メタゲノム解析に限らず、さまざまな実験で出てくるデータ量が増えてきています。すでに実験データの解析にはコンピュータの利用が不可欠になっていますが、今後は並列化による解析の高速化と大規模化がますます必要になってくると考えています。こうしたことに少しでも自分が貢献できたらと考えています。