
Structural analysis of protein in solution by molecular dynamics simulation and small angle X-ray scattering


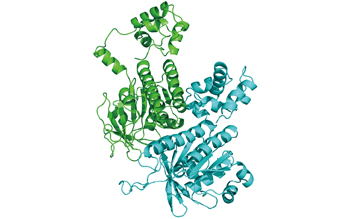
Fig. 1 :Structure of a dimer of Rad51 estimated by MD simulation
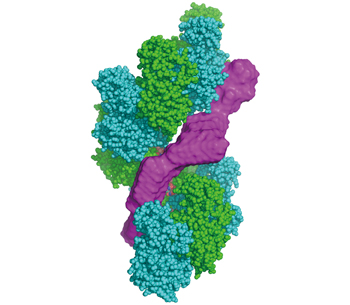
Fig. 2 :Model of Swi5-Sfr1 (magenta) binding to the groove of Rad51 filament in solution
Biopolymers such as proteins exist in solution. A protein in solution does not have a single determined structure, but undergoes structural fluctuations by thermal fluctuation. Therefore, determining the structure of a protein in solution including its structural fluctuations is a key for understanding the in vivo functions of the protein. In this article, two methods for determining (or estimating) the protein structures in solution, namely molecular dynamics (MD) simulation and small angle X-ray scattering (SAXS), are explained by taking our recent study results for example.
MD simulation reproduces behaviors of a protein in solution by solving the equations of motion. With this method, the atomic-level behaviors of a protein can be estimated. We estimated the structure of Rad51 protein filaments in solution, which catalyzes strand exchange in homologous recombination, by performing MD simulation of a dimer as a minimal unit of the filament (Fig. 1).
On the other hand, SAXS irradiates X-rays directly to the protein in solution and determines the general form of the molecule from the scattering pattern. Although the resolution is low, SAXS can measure the protein structure in solution including its structural fluctuations by thermal fluctuation. This time, we analyzed the structure of a protein called Swi5-Sfr1 complex (S/S) in solution, which is an activator of Rad51, by using SAXS, and found that the protein takes an extremely elongated form in solution.
This elongated structure fits well to the groove of the estimated Rad51 filament model (Fig. 2). Therefore, we hypothesized that Rad51 filament is activated by binding of S/S to the groove, and are verifying it.
MD simulation can estimate a structure including fluctuations at an atomic-level resolution, and its confidence factor can be increased by experimental verification. Aiming to verify the results of MD simulation by comparing with the experimental data of SAXS, we developed a theoretical method for calculating the SAXS scattering pattern from the results of MD simulation. We can judge that a result of MD simulation is valid if the SAXS pattern calculated from the MD simulation agrees with the experimental SAXS pattern. This method was applied to a mutant protein Swi5-Sfr1dN177, in which the 177 residues from the N terminus of Sfr1 are lost, to analyze the protein structure in solution including fluctuations at an atomic level.
Today, we are using the method for a DNA-binding protein called a nucleosome. A nucleosome is a huge molecule, and it would require enormous computation, i.e., too much time, to estimate its structure by all-atom MD simulation. Therefore, we are using coarse-grained MD simulation, which calculates the structure by substituting a particle for an amino acid, to analyze the fluctuations in the DNA terminus of the nucleosome and its SAXS scattering pattern.















