
Coarse-grained simulation : Investigation of the molecular mechanism of signaling pathway using CafeMol


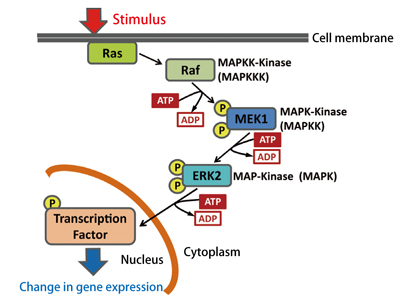
Fig. 1 :MAPK cascade signaling pathway
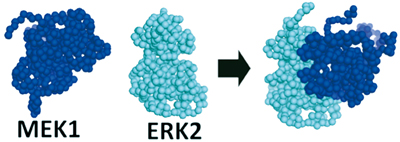
Fig. 2 :Coarse-grained simulation of MEK1 and ERK2 by CafeMol
For cells to adapt to the environment, mechanisms are required for transmitting signals from outside of the cell to the nucleus, amplifying the signals, and expressing a gene and/or changing the activity of a protein. These mechanisms are called a signaling pathway. In our project, we are focusing on mitogen-activated protein kinase (MAPK) cascades, which are a typical signaling pathway model.
For example, in a mammalian MAPK cascade (one of pathways) shown in Fig. 1, a stimulus from outside activates upstream Ras and Raf (MAPKKK), which phosphorylates and activates another protein MEK1 (MAPKK) downstream, and then ERK2 (MAPK). Proteins are thus successively activated (Ras→ Raf (MAPKKK)→ MEK1 (MAPKK)→ ERK2 (MAPK)) until the signal reaches the nucleus. The cascade not only determines the fate of the cell (whether to undergo proliferation, differentiation, or apoptosis, etc.), but is also known to be closely related to canceration (cancer development). Therefore, elucidation of the activation mechanism in signaling pathways is strongly demanded also from a medical point of view. However, the precise molecular mechanism still remains unclear
One of the reasons for the difficulty of understanding the mechanism is that it is challenging to experimentally determine how the upstream protein (for example, MEK1) approaches the downstream protein (for example, ERK2), and forms a complex in terms of atomic-level structure. In our study, we aim to estimate the structure of the MEK1-ERK2 complex and its formation dynamics by using a theoretical approach, and the structures of MEK1 and EKR2 which are already known.
For this purpose we are using CafeMol*, which is a coarse-grained molecular dynamics simulation program developed by our research team. CafeMol assumes each amino acid that constitutes a protein as a bead, and calculates the dynamics. Therefore, the computation cost is significantly reduced compared to all-atom molecular dynamics methods (even a physical event of a millisecond order in a huge system such as a chromatin fiber can be reproduced by making full-scale use of the K computer, a supercomputer). By using our original atomic interaction based coarse grained (AICG) model to represent the interactions (force field) among beads, we can perform more practical calculations than conventional coarse-grained simulation, as our method considers the dependency on the amino sequence and the secondary structures of the proteins.
A complex structure estimated from the coarse-grained simulation using CafeMol is shown in Fig. 2. This structure was compared with the structure estimated from existing docking servers (ClusPro, etc.), and they were found to agree well with each other. CafeMol can also reproduce the time-dependent progress of protein binding, which cannot be performed with a docking server. We are planning to use this advantage to investigate the effects of the presence of a scaffold protein(s) (KSR) and crowded environments within the cell on the dynamics of complex formation, and reveal the molecular mechanism of signal transmission.
Note :
※ H. Kenzaki et al., J. Chem. Theory Comput., 7, pp 1979-1989 (2011) [http://www.cafemol.org/]















