
Various advanced measurement technologies such as cutting-edge sequencers are accelerating refinement and size increases of life system data. Theme 4 “Large-scale analysis of life data” (Leader: Prof. Satoru Miyano, The Institute of Medical Science, The University of Tokyo) aims to construct an infrastructure for cutting-edge, large-scale data analysis optimized for the K computer, understand the complexity, diversity and evolution of life programs, and promote analytical studies on biomolecular networks by using the genome as the standard. We interviewed three researchers who are struggling at the frontline of bioinformatics by utilizing state-of-the-art information processing technologies.
Encounter with bioinformatics
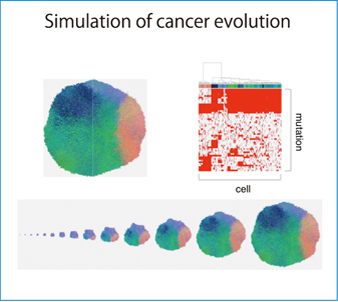
Fig. : Top left: Tumor obtained by simulation calculation
Top right: Profile of genomic mutations
Bottom: Visualized time-historical evolution of a tumor
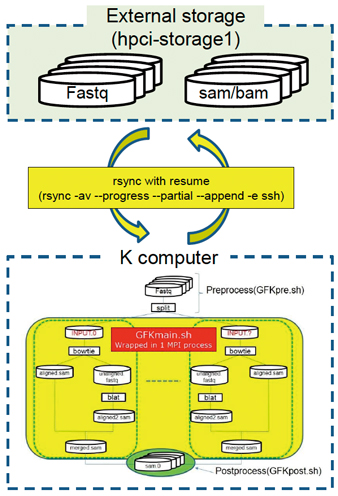
Fig. :The concept of Genomon-Fusion for the K computer, a software for detecting cancer variants on a large-scale and comprehensively at the transcriptomic levels. Various improvements were made to port the program to the K computer such as integration of MPI processes, because multiple MPI processes would lose time due to the MPI barrier mechanism.
●How did you happen to get involved in bioinformatics studies?
Niida: In the undergraduate school, I worked on the molecular biology of cancer. Just when I finished my undergraduate course, the human genome project was completed. The encounter with genomic science aroused my interest in bioinformatics. I was not very good at computers, and so I studied it in my way. After I finished my graduate school, I came to the lab of Prof. Miyano and started to work on the methodology of bioinformatics. One day, Prof. Miyano invited me to the K computer project. I never imagined that I was going to use the fastest computer in Japan, but I’m continuing my studies with help from Dr. Ito and other members.
Ito: I graduated from the school of engineering and majored in computational fluid mechanics. My field is closely related to Field 4, “Industrial Innovation” of the Strategic Programs for Innovative Research. In the field of industrial innovation, computers have been used extensively (Computer Aided Design: CAD), and I was also performing simulation studies using supercomputers. However, technology development in the industrial field is so subdivided or narrow, and studies are conducted on almost every topic. Well, I felt that I wanted to study fundamental science. At that time, at Prof. Miyano’s invitation, I came to this laboratory in April last year. I still do not have a full understanding of biology, and am receiving guidance from Dr. Niida and other members. On the other hand, the members of the laboratory are not yet accustomed to parallelization and optimization of the K computer, and I am supporting that part. We are thus cooperating together.
Kakuta: I was in the department of science during my undergraduate course. Then I went to a graduate school of agriculture to study applied engineering and started to use computer for the first time. In the graduate school, I studied interactions among proteins, and analyzed and estimated three-dimensional structures of proteins. I continued working at the lab for one year and a half after I finished my doctoral course. In the middle of 2011, I moved to the Akiyama Lab and started to participate in the Theme 4 project. I’ve been interested in HPC itself, but I also felt that working on large-scale computation would broaden my studies.
Also working on simulation of cancer
●In Theme 4 project, what actual researches are you conducting?
Niida: The Miyano Lab is carrying out various joint studies with medical practitioners and experimental cancer labs. We receive a huge amount of data from them, which need to be analyzed quickly. One of our ongoing studies is to analyze the data from joint study laboratories by using a software called EEM (Extraction of Expression Module), which extracts expression modules, or genes that are co-expressed, from the mRNA expression data based on gene set information, and identify the transcription program of cancer by combining the analytical results with experimental data. Dr. Ito and other members are helping us to transfer EEM to the K computer. Another study is also simulation of cancer born out of joint studies. While it is well known that the DNA of cancer differs among individuals, we found recently that the DNA is diverse even within a single patient, and differs depending even on the part within a single tumor. It has been found that, in cancer development, the first normal cell accumulates mutation and produces clones that have a mutation(s) different from the mother cell and each other. When we observe a formed tumor, there are many subclones that have mutations in completely different genes. Actually, when we conducted multi-sampling of a colorectal cancer or sampled DNAs from different parts of the tumor in a joint study, we obtained profiles of completely different genes. To answer why such a phenomenon occurs, we will investigate the mechanism of evolution by performing a simulation. Dr. Ito has converted our simulation model so that it runs on the K computer. We are running it while changing the parameters to search for key parameters involved in the development of non-uniformity in a tumor. We are planning to simulate the entire life of a cancer also to determine effective drugs, aiming to use the results in therapeutic strategy.
●Mr. Ito is also involved in the study, engaged in porting the software to the K computer.
Ito: Yes. Not only Dr. Niida, but also most members of the Miyano Lab are not familiar with MPI programs or massively parallel supercomputers such as the K computer. So my first task is to support running of existing software on the K computer. For example, let us assume that Dr. Niida wants to run a certain software on the K computer on a large scale. He tells me his intention, and I analyze the contents of the software, and judge whether the program can be ported to the K computer as it is or not. If it is not possible, I substitute another program for the software, or re-write the program directly to make it run on the K computer. It is also necessary to do parallelization, and I do the work so that Dr. Niida can use the software.
Niida: The K computer is a rather high hurdle for me in some respects. The K computer is designed for persons who have long used HPC, such as those engaged in astronomy and physics. I feel the K computer has parts that are difficult for biologists to use.
Ito: Yes. There are difficult parts. There are various bioinformatics software programs. Each of them is a combination of two or more programs and so, in many cases, they are written in two or more languages instead of one. For example, a pipeline software consists of C language mainly, R statistical language partially, and shell script for connecting C and R parts. Most software needs to resolve the flow before porting. Although there are people in bioinformatics who use supercomputers, there are few who have done parallelization. Therefore, there may be a great barrier to using the K computer without experience of using other HPCI supercomputers.
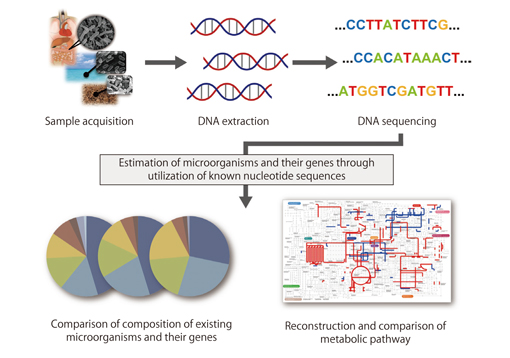
Fig. :Schematic illustration of metagenomic analysis using systematic and functional analyses
Toward ultra-fast large scale analysis
Kakuta: I am mainly developing a metagenomic analysis pipeline using the K computer and also analyzing actual data using the pipeline. Metagenomic analysis is a method for analyzing the genome of the entire group of microorganims within a certain environment. There are various species of microorganisms within an environment, but it is possible to determine what species are present in which part of the environment and their amount by analyzing them as a mixture. It is also possible to investigate how microorganisms interact with the environment by measuring the percentages of the species. However, metagenomic analysis processes a much larger amount of sequence data than genomic sequencing of one microorganism. The project in which I am involved aims to conduct large-scale computation by the K computer to analyze this enormous amount of data. The metagenomic analysis pipeline compares fractions of the genome, which is obtained via a next-generation sequencer, and sequences in the existing databases through a series of processes. The pipeline can identify genes contained in the sample data and analyze their relative abundance based on their functions. A parallelized homology search tool called GHOST-MP plays the central role in the analysis pipeline. Graduate students are writing programs to run on a processor, and I am converting them one by one into a parallelized program that runs on the K computer to enable large-scale analysis. We are aiming at highly precise analysis, but this requires a long computation time. To shorten the time, we are developing efficient and highly parallelized methods, and also increasing the speed of each subprogram. We are trying to develop an ultra-fast, large-scale analysis program by approaching it from both sides.
※Places that are in bold below is part of the long version published only on the Web version.
●Understanding the difficulties of using the K computer for analyzing large-scale data
━What are the difficulties of performing the studies? What are your present problems?
Niida: Well, probably I shouldn’t say this because I’m involved in the HPCI project, but I confess I still need advice from Dr. Ito to handle the K computer. I have to study more about it, not only parallelization and other technical aspects but also its idiosyncrasies. Supercomputers have idiosyncrasies or behave in their peculiar ways, and I really feel that I should get more accustomed to the K computer. For example, when we analyze sequence data, the amount of the data is vast. The supercomputer of the Institute of Medical Science is specialized in processing a large amount of data. On the other hand, the K computer is not, and we have various difficulties. I hope the next-generation supercomputer would be one that can process a large amount of data, such as biological data.
Ito: I am also coming up against this. The most labor-intensive procedure in working with the K computer is preparing the data for processing. These days I spend most of my effort to port a data analysis pipeline called Genomon-Fusion to the K computer. The pipeline is designed to analyze large-scale human specimen data sets, which are available in databases such as CCLE (USA, Broad Institute). The K computer has enough CPU resources to analyze these data in a day. But actually we spent about three months to prepare the data to be analyzed and complete it. We are spending too much time before computation, and are now seeking ways to solve this problem.
Niida: I mentioned the human genome project at the start of this interview. At that time, they took about a dozen years to read the genome of a single person. Today, we can obtain results in several days with advanced sequencers. Data are continuously produced, and we need a large machine like the K computer to analyze them. However, I dare say that the K computer doesn’t have the spec to process a large amount of data smoothly, and that is a problem.
Ito: For example, we can install a software by ourselves. However, there are programs that cannot be installed due to the system architecture, and this needs to be solved on the system side. The data problem we talked about is difficult to solve also from the viewpoint of storage capacity. Data transfer also has difficult points. Particularly, in many cases we need a special software to download data from a database even if it is open for public. Therefore, we download the data to the other supercomputer once, then transfer it to the K computer to analyze, and return the results to that supercomputer. We didn’t imagine that these extra processes were needed, and we just thought we only had to port our software and the K computer would automatically output analytical results. Actually, data handling is also a big problem, and I’m seeking ways to solve it. Computers should be friendly for researchers to use in their work. Supercomputers should be further refined so that not only computer experts but also researchers such as Dr. Niida can fully utilize them. On this point, there are still many problems to be solved.
Kakuta: I also think it is a lot of trouble to transfer data. It is time consuming. Because we cannot reduce the time for the process, there are problems that cannot be solved even if we improve parallelization. I’m struggling to find a way round it.
Niida: Apart from the problems, I actually think it’s very interesting that I’m working with someone like Dr. Ito who is an expert in HPC. I was in life sciences, a completely different field from him, but we are working together on this project. If this project did not exist, I wouldn’t have met Dr. Ito or Dr. Kakuta. It’s fun that people from different fields can sit around the same table, discuss and show ideas, and I also feel a kind of culture shock. This is a very interesting project also in this sense.
Kakuta: I also started to use a supercomputer after I joined this project. Before that, I only used small-scale clusters. At first I didn’t know how to handle the supercomputer and had difficulties, but now I’m feeling that I want to bridge life sciences and computer science. It will certainly be necessary to combine the two. To realize this objective, I must be careful that I don’t end up halfway in both of them.
We want to utilize bioinformatics in medicine
●What do you want to accomplish through this and future studies?
Niida: In the ongoing simulation study, my goals are not only to identify parameters but also to find the principles of evolution and mechanisms that lead to diversity by comparing with experimental data, and to find a new concept of cancer research. To achieve the goals, it is important to improve simulation, but it is also crucial to communicate with in-service medical practitioners to acquire new knowledge, and to feed back experimental data to our own analysis. In the future, I want to make a discovery by using bioinformatics that is useful to medicine.
Ito: My first goals are to develop a pipeline for biologists to analyze a huge amount of data in public and restricted databases in order to acquire biological knowledge, and to carry out computation using the pipeline. Next-generation sequencing technologies are rapidly progressing, thus completely different approaches may be required in several years. To cope with such a situation, I want to develop software programs by merging the characteristics of supercomputers and the requirements of bioinformatics in a sophisticated way.
Kakuta: Not only in metagenomic analysis, but the amount of data is increasing in various kinds of experiments. Computers are already indispensable for analyzing experimental data, but I think that faster and larger-scale analysis by parallelization will be increasingly demanded in the future. I wish I could contribute as much as possible to meeting such a demand.















