
Under this issue, we engage in acceleration and algorithm development required for maximizing efficiency of the post-K molecular dynamics simulation. For drug design application, molecular dynamics simulation covering various target systems is required. In addition to simulation forecasting of protein and ligand binding conditions based on a small system consisting of several tens or hundreds of thousands of atoms, and the dynamics of membrane proteins or protein complexes consisting of a maximum of one million atoms, it is necessary to compute a large-scale system consisting of 10 million atoms or more in the case of a biological system like a virus or cellular environment. To allow the post-K to handle such biological systems, design work is carried out by focusing on both hardware and software to optimize overall computing efficiency, that is, by adopting a co-design methodology. Together with priority issue 1, RIKEN Advanced Institute for Computational Science and Fujitsu Limited are co-designing a molecular dynamics software called GENESIS developed under the initiative of RIKEN. We plan to allow small-and medium-scale systems to implement a number of simulations with different parameters and ligands simultaneously using a post-K, and large-scale systems to challenge complex and large computation limits by using all the nodes of the post-K. By adopting a new molecular dynamics algorithm for drug design application under priority issue 1 in the sophisticated GENESIS, we will attempt to simplify and accelerate the computation of molecular dynamics and the analysis of free energy necessary for drug design through use of the post-K. We will also publish the resulting software and computation so that general users can use them at the same time as the release of the post-K to encourage their use.
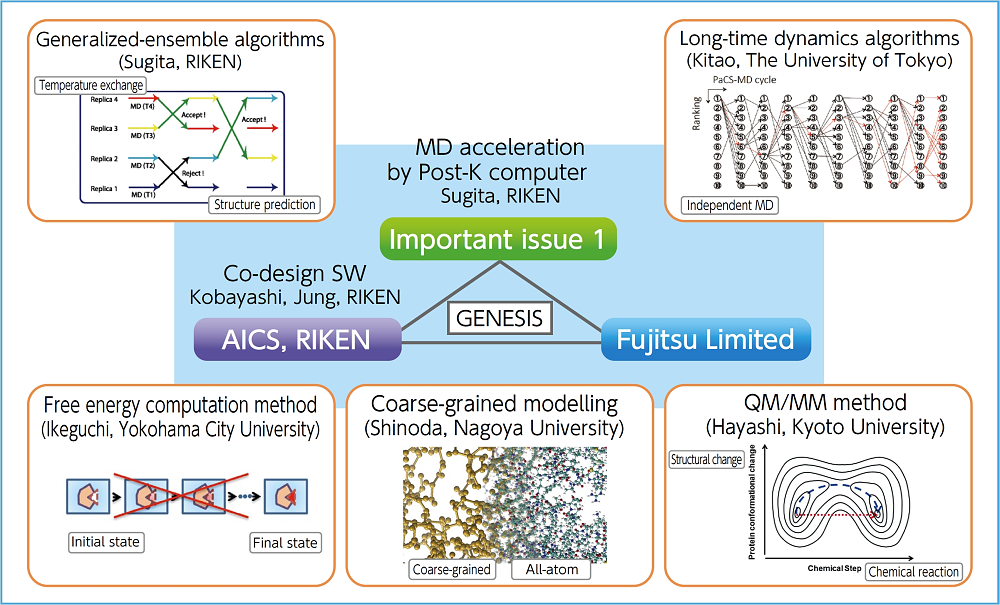
Under Subtheme A, “Post-K MD acceleration and algorithm deepening,” we will make the molecular dynamics software program called “GENESIS” more sophisticated with a view to the post-K concept through the co-design process, and adopt advanced MD algorithms sequentially.

One of the nine priority issues designated under the concept of the post-K computer, “Innovative drug discovery infrastructure through functional control of biomolecular systems,” is now going to start in full swing. In this priority issue, I was appointed as a supervisor for Subtheme B, “Development of next-generation drug design computational techniques.” In Subtheme B, we will work unwaveringly on computing methods for drug design and structural biology, which are not attainable with conventional computing capabilities, by using the sophisticated molecular simulation software and algorithm optimized for the post-K to be developed under Subtheme A, so as to pave the way for the integrated computational drug design system to be developed under Subtheme C. Specifically, first of all, we set a goal to control dynamic molecular functions by focusing on motions and structural changes of drug design target proteins. Computing methods for conventional computational drug design often treat the structures of target proteins as firm and immovable, in accordance with the lock-and-key model. However, actually, the structures of proteins are very soft and often change due to the binding of compounds such as pharmaceuticals. In this issue, we try to understand and control the motions and structural changes of target proteins by capitalizing on the superior computing capabilities of the post-K. The next issue is to control the interactions between proteins, and those between nucleic acids and proteins. In recent years, bio-pharmaceuticals containing proteins as medicinal chemicals such as antibody drugs also have been developed. Such proteins and nucleic acids are much more complex than low-molecular weight compounds, and their steric structures are flexible. Therefore, it is necessary to conduct large-scale computation using the post-K in order to compute the interactions between molecules in detail. Our issues also include the entire computation of virus capsids and computation targeted at the cellular environment where a plural number of biological supermolecules are intricately entwined. Actually, these targets are supergiant systems, which could be computed only by the world's leading supercomputers. This would be an unprecedented challenge. As described so far, this is already an attractive research theme only in terms of computation. However, its value will be increased by closely coordinating with experimental system measurements. In this issue, we plan to conduct research activities in cooperation with the most advanced structural biology experiment facility SPring8 and NMR facilities. We sincerely hope that we can implement unprecedented computations for drug design using the post-K.
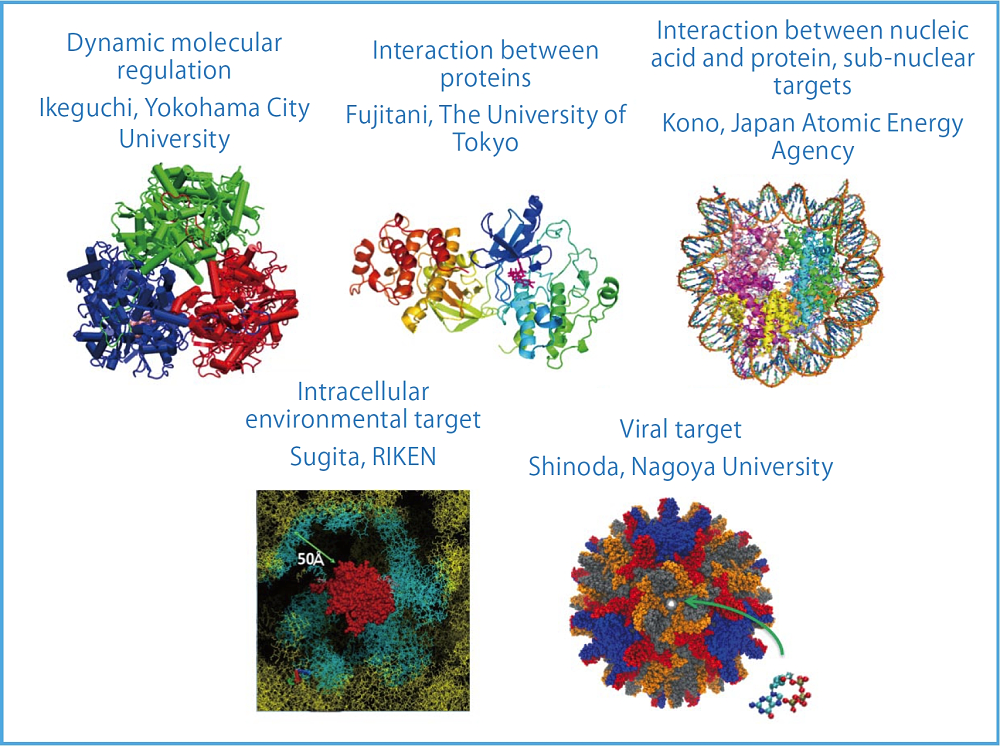
Outline of Subtheme B “Development of next-generation drug design computational techniques.” It consists of the subjects “dynamic molecular regulation,” “control of interaction between proteins ,” “control of interaction between nucleic acid and protein,” “sub-nuclear environmental target,” “intracellular environmental target,” and “viral target.”
![]()
Priority issue (1) : Innovative drug discovery infrastructure through functional control of biomolecular systems
Subtheme A : Advancement of MD and associated algorithms by post-K computer
Subtheme B : Development of next-generation drug design computational techniques
Priority issue (2) : Integrated computational life science to support personalized ized and preventive medicine

 |
 |
 |
 |
 |
|---|










