
シミュレーションで見る分子認識のメカニズムと薬のデザイン

東京大学 先端科学技術研究センター
山下 雄史
![]()
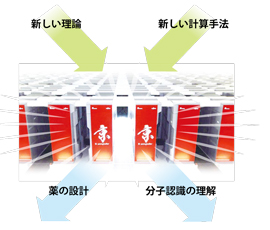
我々は、「スーパーコンピュータを使った薬づくり」の研究をおこなっています。病気の原因となるタンパク質に強く作用する分子を探すために、大規模な計算をスーパーコンピュータでおこない、解析して薬づくりに応用しています。また、そのための新しい解析手法や理論の開発をしています。創薬応用シミュレーションという分野の内容は、基礎的・学問的な側面から応用的・技術的な側面まで多岐に渡ります(図1)。
学問的な側面から見れば、薬のデザインは「分子を認識するメカニズムを知る」ということになります。多くの薬は、病気の原因となるタンパク質の機能を抑えることで、薬としての役割を果たします。もし薬が目標のタンパク質だけでなく、いろいろなタンパク質の機能に影響を及ぼすとしたら、どうなるでしょうか?これは致命的な副作用を引き起こす原因になってしまいます。したがって、薬は他のモノには目もくれず、目標のタンパク質だけを「認識」して攻撃できなければならないのです(図2)。
分子がどれくらい強く目標のタンパク質を認識できるかを表す物理量の1つに「結合自由エネルギー」というものがあります。結合自由エネルギーの値が大きい分子は、標的のタンパク質を強く認識して攻撃できるため、薬として非常に有望なものになります。我々は、この結合自由エネルギーを正確に予測することができれば、薬開発の大きな武器になると考えています。そこで、我々は藤谷ら[1]が提案している結合自由エネルギー計算法を用いて、実際のがんの原因タンパク質に応用を試みています。この手法では、1つの分子と標的タンパク質の結合自由エネルギーを評価するだけでも大きな計算資源を必要とします。薬として適した分子を探すためには、多くの分子を計算する必要がありますが、従来のコンピュータでは非常に難しいことでした。それが、スーパーコンピュータ「京」の登場により、ある程度、実行可能な時代に入りました。
実際に、「京」を用いた超大規模計算を始めてみると、想定外の問題に直面したりもしましたが、徐々に新しい知見が蓄積されてきています。こうした知見を積み上げていくことで、分子認識のメカニズムを「より深く」、「より正しく」理解していくことができると思っています。分子認識のメカニズムが理解できれば、従来の経験的な薬づくりを脱却し、もっと論理的な方法で簡単に薬をつくれる時代が来るのではないかと考えています。
分子シミュレーションの利点は、直接には見ることができない水中のタンパク質の様子を、高精度な顕微鏡を覗くように見ることを可能にする点でしょう。さらに面白いのは、本当は起こり得ないバーチャルな状況をつくることが可能な点です。実は、このバーチャルな状況を上手く活用することで、現実の物理量をより高速に計算する工夫ができるのです。実際、我々の結合自由エネルギー計算の方法もこうしたアイデアを使っています。計算機だからこそ可能な方法を開発していくことも、この分野の大きな魅力です。
【参考文献】
[1] H. Fujitani, Y. Tanida, and A. Matsuura: Phys. Rev. E ,79 021914 (2009), H. Fujitani, K. Shinoda, T. Yamashita, and T. Kodama: J. Phys. Conf. Ser . 454 012018(2013)
 |
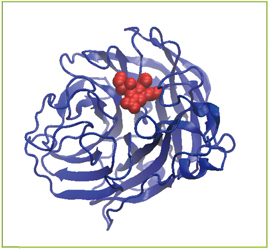 |
図1:創薬応用シミュレーション分野のイメージ |
図2:インフルエンザウィルスのタンパク質(青色で表示)に結合しているリレンザという薬(赤色で表示) |